10.3 Характеристика основних класів ферментів
Окиснювально-відновні ферменти (оксидоредуктази)
Під час процесів переробки рослинної сировини разом з гідролітичними використовуються і окисно-відновні ферменти. Оксидоредуктази застосовуються переважно на завершальних стадіях переробки рослинних субстратів з метою збереження харчових і смакових переваг харчових продуктів або модифікації органолептичних властивостей.
Варто пам'ятати, що представники усіх класів ферментів в тих або інших кількостях є присутніми в рослинній сировині, що переробляється, і ферментативна активність сировини неминуче впливає на хід технологічних процесів переробки. Цей вплив може бути як корисним, так і шкідливим. У другому випадку необхідно регулювати ферментативну активність в сировині шляхом його попередньої обробки або введення технологічно допустимих компонентів. Прикладами небажаних реакцій є окиснювальні перетворення органічних сполук в рослинній сировині, такі як потемніння подрібнених овочів і фруктів під дією тірозинази і дифенолоксидази, а також згіркнення жирів, спровоковане ліпоксигеназою.
Глюкооксидаза (КФ. 1.1.3.4). Фермент є флавопротеїдом, в якому білок сполучений з двома молекулами ФАД. Він окислює глюкозу з утворенням в кінцевому випадку глкюконової кислоти і має практично абсолютну специфічність по відношенню до глюкози. Сумарне рівняння має наступний вигляд:
Глюкоза + Н2О + О2 = глюконова кислота + Н2О2
Представлений вище процес насправді відбувається в декілька стадій:
1-я стадія:
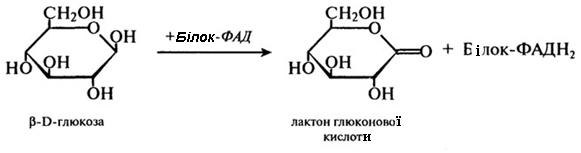
2-я стадія:
3-я стадія:
Білок-ФАД○Н2 + О2 → Білок-ФАД + Н2О2
4-я стадія:
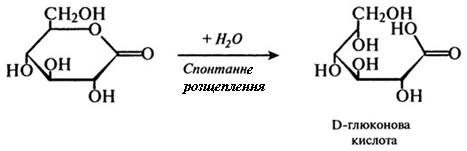
На першому етапі цієї реакції відбувається видалення двох атомів водню у першого вуглецевого атома глюкози. Таким чином утворюється відновлений флавіновий фермент і лактон глюконової кислоти. Далі відновлений фермент реагує з киснем повітря, і утворюється пероксид водню. Токсичний пероксид водню розщеплюється каталазою на кисень і воду, а β-О-глюконо-δ-лактон піддається спонтанному розщепленню з приєднанням води, внаслідок чого утворюється глюконова кислота.
У харчовій технології глюкозооксидазу використовують спільно з каталазою , оскільки необхідно розкладати пероксид водню, що утворюється на першій, ферментативній стадії окиснення глюкози глюкозооксилазою.
Високоочищені препарати глюкозооксидази отримують з цвілевих грибів роду Aspergillus і Penicillium. Вони мають приблизно однакову молекулярну масу ≈150 кД, ІЕТ – 4,2...4,3 і оптимум рН 5,6, стабільний в зоні рН 3...7, за температури до 50ºС. Інгібітори – іони Гідраргіруму, Купруму, стабілізатори – іони Кальцію й амонію.
Останніми роками глюкозооксидаза отримала широке застосування. Завдяки винятковій специфічності препарати глюкозооксидази застосовуються як аналітичний засіб для кількісного визначення глюкози.
Окрім цього, препарати глюкозооксидази знайшли застосування в харчовій промисловості для таких випадків:
– для видалення слідів глюкози, що є необхідним під час обробки харчових продуктів, якість і аромат яких погіршуються через те, що в них містяться відновлюючі цукри; наприклад, в отриманні з яєць сухого яєчного порошку (мається на увазі реакція Майяра, так як глюкоза під час висушування і зберігання яєчного порошку, особливо за підвищеної температури, легко вступає в реакцію з групами амінів амінокислот і білків в наслідок чого порошок темніє, і утворюється ряд речовин з неприємним смаком і запахом);
– для видалення слідів кисню, що є необхідним під час обробки продуктів, в яких тривала присутність невеликих кількостей кисню призводить до зміни аромату і кольору (пиво, вино, фруктові соки, майонез). Внесення пакетиків, що містять суміш води, глюкози, ферменту і буферу, сприяє видаленню кисню з повітряного простору. В усіх подібних випадках у ферментну систему включають каталазу, яка розкладає Н2О2, що утворюється під час реакції глюкози з киснем. Цей метод знайшов широке застосування в США для видалення кисню з банок з сухим молочним порошком.
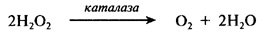
Каталаза (КФ.1.11.1.6) – універсальний фермент органічного світу, що приймає участь в завершальних стадіях процесу окиснення. Він каталізує розкладання пероксиду водню відповідно до наступної реакції:
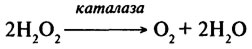
Таким чином, фермент окислює одну молекулу пероксиду водню до кисню з одночасним відновленням іншої молекули пероксиду водню до Н2О.
Фермент здатний також каталізувати окиснення спиртів в альдегіди, що пов'язане з розкладанням пероксиду водню :
Каталаза + Н2О2 →Каталаза○Н2О2
Каталаза○Н2О2 + С2Н5ОН →Каталаза +2Н2О + СН3СНО
Такий тип реакції характерний для середовищ з низьким вмістом пероксиду водню.
Каталаза відноситься до групи гемопротеїнових ферментів. Містить 0,009% Феруму у вигляді гемінового угрупування або 4 атоми на одну молекулу ферменту. Молекулярна маса ферментів, виділених з різних об'єктів (дріжджів, рослинних і тваринних тканин, мікроорганізмів) – в межах від 225 кД до 250 кД. Вони мають істотні відмінності в оптимумі рН (від 2 до 9), в термо- і рН-стабільности. Фермент інгібується ціанідом (оборотно), фенолами (оборотно лише в слабкій формі), лугом і сечовиною (безповоротно). Функцією каталази в живому організмі є захист клітини від згубної дії пероксиду водню.
У харчовій промисловості використовуються препарати каталази мікробного походження, що виділяються з грибів пеніцилів. Каталаза Pen.Vitale має широкий рН-оптимум – від 4 до 9, стабільна за температури до 650С.
Каталаза знаходить своє застосування в харчовій промисловості в процесі видалення надлишку Н2О2 під час обробки молока в сироварінні, де остання використовується як консервант; а також спільно з глюкозооксидазою застосовується для видалення кисню і слідів глюкози.
Враховуючи пов'язаність дії глюкозооксидази і каталази, ці ферменти виділяють не лише як індивідуальні, але і у вигляді комплексних препаратів. Гриби пеніцили і аспергіли мають здатність продукувати значні кількості обох ферментів.
Пероксидаза (КФ.1.11.1.7). Пероксидаза каталізує окиснення органічних сполук за допомогою пероксиду водню і органічних пероксидів, що наприклад, утворюються з ненасичених жирних кислот, каротиноїдів. Фермент переносить кисень від молекули субстрату до пероксиду. Субстратами пероксидази служать різні сполуки – феноли (пірокатехін, пірогалол, гідрохінон, резорцин, гваякол), ароматичні кислоти (бензойна, саліцилова, галова), аскорбінова кислота, анілін, толуїдин, нітрит й інші сполуки.
Сумарно реакцію можна представити так:
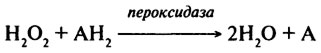
Механізм реакції ґрунтується на утворенні комплексів фермент-донор і двох одновалентних ступенів окислення:
Пероксидаза + Н2О2
Комплекс I + АН2
Комплекс II + АН = Пероксидаза + А
Більшість субстратів пероксидази – феноли. Під дією ферменту вони окиснюються до хінонів, які самі по собі є сильними окисниками. Хінони є схильними до полімеризації, в результаті чого утворюються темнозабарвлені сполуки.
Пероксидаза є присутньою в кожній рослинній клітині. Цей фермент приймає участь в циклах фотосинтезу і дихання, відіграє істотну роль в захисті рослин від інфекційних захворювань. Спостерігається позитивна кореляція між ступенем ушкодження рослини, концентрацією фенолів і активністю пероксидази. Тому під час переробки інфікованої рослинної сировини рівень окиснювальних процесів в цілому є вищим, ніж під час переробки неушкодженого матеріалу.
Пероксидаза – двокомпонентний фермент, що складається з білку глікопротеїну і гемінового компоненту (активний центр). Фермент містить один атом Феруму на молекулу, має молекулярну масу від 22 до 44 кДа, білкова частина на 43% є спіралізованою.
Пероксидаза представлена в рослинах набором ізоферментів, число їх у одного виду може складати від 3 до 42. З різних видів тютюну виділили 33 ізоферменти пероксидази. Наявність ізоферментів розширює межі функціонування пероксидази, вона проявляє активність в зоні рН 3...14. В результаті зміни зовнішніх умов і гомеостазу рослини змінюється ізоферментний склад пероксидази, що є механізмом пристосуванням.
Найбільш активна пероксидаза виділена з коренів хріну. Її молекулярна маса дорівнює приблизно 40 кД, ІЕТ – 7,2, стійка за рН від 4 до 12; оптимум рН для пероксидази хріну дорівнює 7; за рН від 6 до 8 зберігається 70% його активності.
Регуляція дії пероксидази здійснюється за допомогою іонів металів – Мангану, Цинку, Купруму, Кальцію та ін. Їх присутність впливає на співвідношення власне пероксидазної, оксидазної і оксигеназної активності.
Інгібіторами пероксидази є ціаніди і хелати. За допомогою хелатів, таких як ЕДТА, лимонної кислоти та її солей, можна запобігти окиснювальним реакціям, що відбуваються в рослинній сировині під дією пероксидази.
о-Дифенолоксидаза (КФ.1.14.18.1). Цей фермент є відомим також і під іншими назвами: поліфенолоксидаза; тирозиназа; фенолаза; катехолаза. Він каталізує окиснення дифенолів, поліфенолу, монофенолів, дубильних речовин за допомогою кисню повітря.
Типова реакція, що каталізується о-дифенолоксидазою, має вигляд:
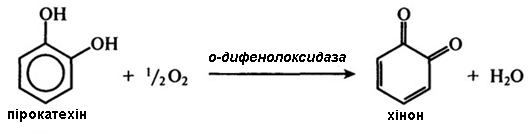
Найменш стійкими до окиснення є о-дифеноли, у яких гідроксильні групи розташовані біля сусідніх вуглецевих атомів.
Молекула ферменту має четвертинну структуру і молекулярну масу приблизно 34000 Д. о-дифенолоксидаза – купропротеїд. Вміст Купруму – 0,2%, або один атом Сu на одну молекулу ферменту. Зона оптимальної активності – між рН 5,0...7,0.
Дифенолоксидаза – універсальний рослинний фермент, присутній в усіх органах і тканинах. В залежності від того, з якого джерела виділений фермент, здатність його до окиснення різних фенолів є різною. Більше того, навіть в одному і тому ж об'єкті о-дифенолоксидаза може міститися у вигляді різних молекулярних форм (ізоформ), що відрізняються за здатністю до окислення різних фенолів.
З дією цього ферменту пов'язано утворення темнозабарвлених сполук – меланінів в результаті окиснення киснем повітря амінокислоти – тирозину. Під дією дифенолоксидази рослинні феноли окиснюються спочатку в хінони, які, конденсуючись, перетворюються на меланіни. Колір меланінів залежить від їх молекулярної маси: чим більшою є молекулярна маса, тим темнішим є забарвлення, в міру збільшення молекулярної маси колір змінюється від рожевого до чорного. Потемніння зрізів картоплі, яблук, грибів, персиків й інших рослинних тканин в більшій мірі або повністю залежить від дії о-дифенолоксидази. У харчовій промисловості основний інтерес до цього ферменту зосереджений на запобіганні ферментативному потемнінню, яке має місце під час висушування плодів і овочів, а також у виробництві макаронних виробів з борошна з підвищеною активністю о-дифенолоксидази. Ця мета може бути досягнута наступними шляхами:
– теплова інактивація ферменту (баланширування), інактивація за допомогою підкислення (за рН<3 фермент є нестабільним);
– додавання інгібіторів (аскорбінова кислота, NaHSO3, SO2, NaCl), за присутності яких відбувається відновлення о-хінонів у феноли і пригнічується процес конденсації хінонів;
– зв'язуванням субстрату за допомогою метилування.
Гарні результати дає витримка матеріалу, що пройшов механічну обробку (очищення, різання), в слабких розчинах лимонної кислоти, яка має хелатну дію: зв’язує каталітично активні іони Купруму, що в свою чергу, інактивує дифенолоксидазу. Своєчасне занурення рослинного матеріалу у воду (а ще краще – в злегка підсолену воду) також є досить ефективним. Процеси окиснення розвиваються з часом, тож якщо матеріал негайно залити водою, роз’єднавши тим самим з киснем повітря, можна запобігти потемнінню.
Позитивна роль ферменту проявляється в деяких ферментативних процесах: наприклад, під час ферментації чаю. Окислення дубильних речовин чаю під дією о-дифенолоксидази призводить до утворення темнозабарвлених і ароматичних сполук, які визначають колір і аромат чорного чаю.
Ліпоксигеназа (КФ.1.13.11.12) каталізує окиснення поліненасичених жирних кислот (лінолеву, ліноленову, арахідонову), та їх естерів киснем повітря з утворенням високотоксичних гідропероксидів. Ненасичені жирні кислоти перетворюються на гідропероксиди, таким чином змінюється положення подвійного зв'язку Нижче наведена реакція, що каталізується цим ферментом:
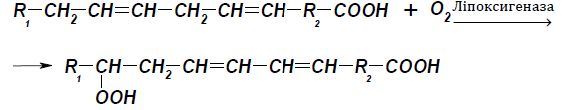
Можливим є утворення і циклічних пероксидів за наступною схемою:
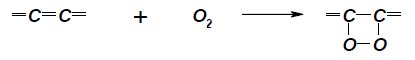
Проте основна кількість жирних кислот перетворюється на гідро-пероксиди, що мають сильні окиснювальні властивості, і саме на цьому засноване використання ліпоксигенази в харчовій промисловості.
Гідропероксиди жирних кислот – активні окиснювальні агенти, здатні окиснювати ненасичені жирні кислоти, каротиноїди, хлорофіл, амінокислоти, аскорбінову кислоту. Таким чином, дія ліпоксигенази ініціює цілий ряд різних окиснювальних реакцій в рослинній сировині.
Висока ліпоксигеназна активність знайдена в насінні, що перебуває в спокої таких культур як бобових, льону, в зародках злаків. У насінні соняшнику, рапсу, конопель, горіха активність ферменту є невеликою, вона зростає під час проростання насіння.
Ліпоксигеназа – залізовмісний глобулін. Молекулярна маса рослинних ліпоксигеназ – в межах 67...108 кД, оптимальний рН 6,2 ...7,5, температура 20...40ºС.
Найбагатшим джерелом ферменту є борошно соєвих бобів. У зерні пшениці активність ліпоксигенази коливається в значних межах і є сортовою ознакою. Крім того, активність ліпоксигенази пов'язана з показником життєздатності зерна. Вона закономірно знижується зі зниженням схожості зерна і може бути біохімічним тестом життєздатності насіння. Значна частина ліпоксигенази пшениці є міцно зв'язаною з білками клейковини і звільняється під час обробки комплексу клейковини розчином відновленого глутатіону.
Ліпоксигеназі належить важлива роль в процесах дозрівання пшеничного борошна, пов'язаних з поліпшенням її хлібопекарських властивостей, продукти окиснення жирних кислот, що утворюються під дією ферменту, здатні викликати зв'язане окиснення ряду інших компонентів борошна (пігментів, SH- груп білків клейковини, ферментів та ін.). Таким чином відбувається освітлення борошна, зміцнення клейковини, зниження активності протеолітичних ферментів і інші позитивні зміни.
Утворення гідропероксидів жирних кислот – початкова стадія їх деградації, в процесі якої утворюються леткі речовини, що створюють характерний запах згірклих олій, борошна, круп за їх тривалого зберігання.
Для запобігання негативним наслідкам дії ліпоксигенази в продуктах, що зберігаються, роблять різні заходи. Щоб уникнути згіркнення борошна під час помелу зерна відділяють фракцію зародків – найбільш багату ліпоксигеназою частину зерна. Під час розфасування рослинних олій і жировмісних продуктів за можливості виключають попадання кисню або використовують спеціальні пакувальні матеріали, на які нанесений фермент глюкозоксидаза, її субстрат глюкоза і буферні солі (система, що поглинає кисень в реакції окиснення глюкози в глюконову кислоту).
Широке застосування знайшли різні антиоксиданти – токофероли, каротиноїди, янтарна і фумарова кислоти, флавоноїди (морин, кемпферол, мірицетин, кверцетин, дегідрокверцетин та ін.), аскорбінова кислота, рослинні екстракти, що містять комплекс різноманітних антиоксидантів.
Гідролітичні ферменти
Гідролітичні ферменти є найважливішим класом ферментів, що використовуються у переробці харчової сировини. Основною продукцією ферментної промисловості є препарати гідролаз, таких як амілази, протеази, пектинази, целюлази. Гідролітичні ферменти використовуються переважно в початковій, найбільш трудомісткій стадії переробки органічної сировини, коли необхідно розщепити структурні або запасні полімери, фрагменти яких далі піддаються трансформації під дією ферментів інших класів.
Субстрати гідролітичних ферментів – полісахариди, білки, ліпіди, нуклеїнові кислоти й інші природні сполуки, що містять "ангідридові" зв'язки – складають основну масу органічної матерії на планеті. Гідроліз – необхідна стадія кругообігу цих сполук в природі, тому гідролітичні реакції, як хімічні, так і ферментативні, відбуваються у великих масштабах в природних умовах.
Реакція гідролізу відбувається за наступним рівнянням:
RR1+H–OH D RH+R1OH
В процесі ферментативного гідролізу відбувається утворення фермент-субстратного комплексу, який зазнає внутрішньомолекулярного перегрупування під впливом активного центру ферменту. Каталізований розрив ангідридового зв'язку субстрату призводить до виділення з фермент-субстратного комплексу одного з продуктів реакції. Другий продукт виділяється після перегрупувань, які пов'язані з приєднанням молекули води.
Клас гідролаз – третій клас в номенклатурі ферментів – включає 11 підкласів. Ферменти підкласу 3,1 розщеплюють естерні зв'язки; 3.2 – глікозидні зв'язки; 3.3 – естерні зв'язки; 3.4 – пептидні зв'язки; 3.5 – зв'язки C–N, відмінні від пептидних; 3.6 – кислотноангідридні зв'язки; 3.7 – зв'язки С–С; 3.8 – галоїдалкілні зв'язки; 3.9 – зв'язки P–N; 3.10 – зв'язки S–N; 3.11 – зв'язки С–Р.
Найбільше застосування в процесах промислового біокаталізу знайшли гідролази підкласів 3.1, 3.2 і 3.4.
Естерази (КФ.3.1)
Цей підклас включає велике число ферментів (приблизно 150). Ферменти підкласу 3.1 розщеплюють естерні зв'язки. Для естераз характерна відносна групова субстратна специфічність, тобто здатність гідролізувати естерні зв'язки між радикалами різного типу. Естерази розщеплюють моно-, ди-, тригліцероли й інші сполуки, що містять естерний зв'язок. Швидкість розщеплення залежить від структури субстрату. Естерази поділяють на сім підкласів: ферменти, що діють на естери карбонових кислот (3.1.1); естерази тіолових естерів (3.1.2); гідролази фосфорних моноестерів або фосфатази (3.1.3); гідролази фосфорних діестерів (3.1.4); гідролази моноестерів олігофосфорних кислот (3.1.5); сульфатази (3.1.6); естерази моноестерів дифосфорних кислот (3.1.7).
Найбільш важливими з точки зору участі в різних біохімічних процесах, що мають місце під час зберігання й переробки харчової сировини, є ферменти підкласу 3.1.1, що діють на естери карбонових кислот.
Ліпаза (КФ.3.1.1.3) або тріацилгліцерол-ліпаза широко розповсюджена в природі і відіграє важливу роль в процесах, що відбуваються під час переробки і зберігання харчових продуктів. До таких ліпаз належать: ліпази рослинного походження (ліпаза рицини, пшениці й інших злаків), тваринного (панкреатична ліпаза, ліпаза молока) і мікробного (бактерійні і грибні ліпази).
Ліпази є універсальними ферментами що використовуються для перетворення ліпідів. Субстратами ліпаз є гліцериди й інші естери. Ліпази більше за інших представників класу гідролаз мають здатність каталізувати різні типи реакцій, а саме, наступні типи:
|
1 – Гідроліз естерів |
R–COO–R1 + H2O→R–COOH +R1–OH |
|
|
2 – Синтез естерів |
R–COOH +HO–R1→R–COO–R1 +H2O |
|
|
3 – Трансестерифікація |
|
|
|
|
3.1 Ацидоліз |
R–COO–R1+ R2–COOH→R2–COO–R1 +R–COOH |
|
|
3.2 Алкоголіз |
R–COO–R1 + HO–R1→R–COO–R2 + HO–R1 |
|
|
3.3 Інтерестерифікація |
R–COO–R1 + R2–COO–R3→R–COO–R3+ R2–COO–R1 |
|
|
3.4 Аміноліз |
R–COO–R1 + H2N–R2→R–CO–NH–R2 +HO–R1 |
Гідролітичне розщеплення жирів ліпазами відбувається з помітною швидкістю в середовищах з концентрацією води не менше 1%. Реакція відбувається в гетерогенному середовищі на межі поділу фаз ліпіди-вода, швидкість її залежить від ступеня дисперсності субстрату.
Зазвичай ліпази каталізують реакцію розщеплення тригліцеридів згідно з наведеним нижче сумарним рівнянням:
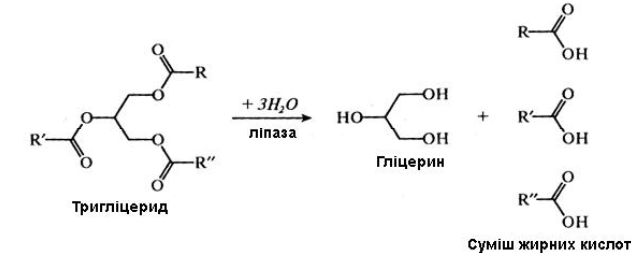
Ліпази проявляють специфічність відносно оптичних ізомерів естерів (стереоспецифічність), позиційну, гліцеридну і жирокислотну специфічність.
За позиційною специфічністю ліпази поділяють на дві групи: позиційно неспецифічні, які звільняють в процесі гідролізу тригліцеридів жирні кислоти з усіх трьох позицій, і 1,3-специфічні.
Більшість відомих ліпаз переважно гідролізують естерний зв'язок біля С1 і С3 гліцеролу.
За тривалого гідролізу гліцеридів 1,3-специфічні ліпази здатні відщепити жирні кислоти з усіх положень, оскільки 2-моногліцериди і 1,2-дигліцериди, як менш конформаційно стабільні, мимоволі ізомеризуються в 1-моногліцериди і 1,3-дигліцериди.
Жирнокислотна специфічність ліпаз виражається в перевазі до жирних кислот певної довжини ланцюга. В цілому ліпази легко відділяють жирні кислоти середньої довжини.
Гліцеридна специфічність виражена не в усіх ліпаз. Фермент з Pen.cyclopium гідролізує моногліцериди і дигліцериди і практично не діє на тригліцериди.
Ліпази різного походження сильно відрізняються одна від одної за специфічністю дії, спорідненістю до різних субстратів, розчинністю, оптимуму рН й іншим властивостям. Так, наприклад, ліпаза насіння рицини є нерозчинною у воді, має оптимум рН 4,7...5,0; панкреатична ліпаза – розчинна, й оптимум рН її дії знаходиться в слаболужному середовищі. Ліпаза пшеничних зародків також відрізняється від ліпази рицини. Вона є розчинною у воді і має рН оптимум 8,0.
Для мікробних ліпаз-ферментів з молекулярною масою 30...55 кДа, оптимальна температура дії не перевищує 65°С, оптимальний рН більшості досліджених ліпаз знаходиться в слабокислій і нейтральній зоні. Бактерії роду Pseudomonas продукують лужні ліпази і ліпази з широкою зоною рН-оптимуму.
Зернова ліпаза приймає участь в процесі псування зернових продуктів під час зберігання. Особливо це стосується продуктів, що містять підвищену кількість жиру, наприклад, вівсяного борошна або крупи, пшона. Накопичення вільних жирних кислот під дією ліпази (зростання кислотного числа жиру) є ознакою погіршення якості продукту. Вільні жирні кислоти, особливо ненасичені, легко піддаються окисненню під впливом різних факторів: ліпоксигенази, теплової обробки, кисню повітря, сонячного світла та ін. Таким чином, ліпази можуть ініціювати процес згіркнення й обмежувати терміни зберігання харчових продуктів.
Пектинестерази, ПЕ (КФ.3.1.11) каталізують гідроліз естерних зв’язків в молекулі розчинного пектину, а саме, відщеплення метильних груп від поліметилгалактуронової кислоти з утворенням метанолу і частково диметоксильованої полігалактуронової кислоти.
Процес відбувається згідно з наступною схемою (стрілками показана дія ферменту):
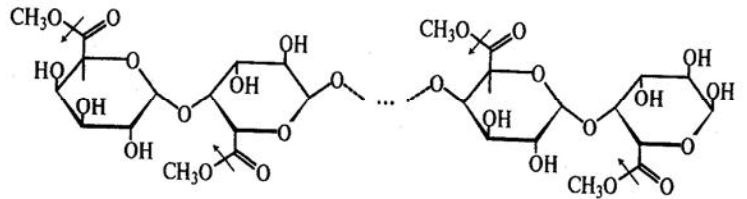
ПЕ деестерифікує пектини на 60...70%. Зі зниженням ступеня естерифікації субстратів зменшується спорідненість ферменту до них, і процес гідролізу не проходить до кінця. ПЕ переважно діє на великі молекули, метоксильовані олігоуроніди розщеплюються повільніше.
Пектинестерази синтезуються вищими рослинами, мікроскопічними грибами, дріжджами і бактеріями.
Пектинестерази проявляють максимальну активність в інтервалі рН 4,4...8,0, у деяких мікроскопічних грибів за рН 2,5. Оптимальна температура дії 30...40°С. Пектинестераза входить в комплекси пектолітичних ферментних препаратів мікробного походження.
Желююча здатність пектину залежить від ступеня метоксилювання або ступеня естерифікації, тому дія пектинестерази з відщеплення метоксильних груп призводить до зниження желюючої здатності і супроводжується падінням в'язкості. На цьому, очевидно, і ґрунтується застосування цього ферменту для освітлення плодових соків і вина.
Зазвичай комплексні препарати пектолітичних ферментів, що використовуються з цією метою, отримують з різних цвілевих грибів, і передусім з A.Niger
Гідролази глікозидів або Глікозидази (КФ.3.2)
Цей підклас включає приблизно ста ферментів з різною специфічністю дії, що здійснюють гідроліз оліго- і полісахаридів; деякі ферменти цього типу здатні здійснювати трансферазні реакції – переносити глікозидні залишки на оліго- і полісахариди, нарощувати полісахаридні ланцюжки.
Глікозидази є стереоспецифічними. Вони гідролізують глікозидні зв'язки певної просторової конфігурації (α чи β), але не обох одночасно. Прикладами можуть служити α-амілаза, α-глюкозидаза, глюкоамілаза, що гідролізують виключно α-глікозидні зв'язки, і β-глюканаза, целобіаза, лізоцим – гідролізують β-зв'язки.
Менша вибірковість проявляється до різних видів α- або β-глікозидних зв'язків. Так, глюкоамілаза розщеплює α-1,4 і α-1,6-зв'язки; глюканаза бацил – β-1,3 і β-1,4-зв'язки;
Глікозидази є специфічними відносно довжини ланцюга гідролізованих полімерних субстратів. Так, глюкоамілаза цвілевих грибів переважно гідролізує високомолекулярні полімери α-1,4-зв'язаної глюкози, а аналогічний фермент – α-глюкозидаза дріжджів каталізує гідроліз того ж зв'язку в олігосахаридах, але не в крохмалі.
Основною формою запасних вуглеводів в насінні і бульбах рослин є крохмаль. Ферментативні перетворення крохмалю лежать в основі багатьох харчових технологій.
Група ферментів, що гідролізують крохмаль (амілолітичних), включає: α-амілазу, β-амілазу, глюкоамілазу, α-глюкозидазу, ізоамілазу, пулуланазу.
α-Амілаза (α-1,4-глюкан-4-глюканогідролаза, КФ.3.2.1.1) гідролізує α-1,4-глікозидні зв'язки в крохмальних полісахаридах і глікогені, вона є ферментом ендотипу – гідролізує α-1,4-глікозидні в середині молекули крохмалю, розриваючи зв’язок між першим атомом Карбону і Оксигеном, що з’єднує цей атом Карбону з сусідньою молекулою глюкози). Це наочно демонструє наступна схема (стрілками показана дія ферменту):
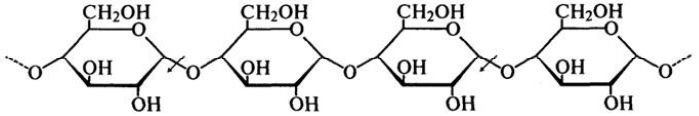
Швидкість з якою α-амілази гідролізують глюкани з різним ступенем полімеризації знижується в міру зниження цього ступеня полімеризації. Амілоза – лінійна фракція крохмалю, гідролізується швидше, ніж амілопектин, що має розгалужену структуру.
Швидкість гідролізу α-амілазою залежить від виду і стану крохмалю (нативний або клейстеризований), а також від ефективності самих амілаз.
α-Амілази виявлені у тварин (у слині і підшлунковій залозі), в рослинах (проросле зерно пшениці, жита, ячменю), вони виробляються цвілевими грибами і бактеріями.
Багато амілаз містять кальцій (один атом на молекулу ферменту). Роль кальцію полягає в тому, що він стабілізує вторинну і третинну структури молекули α-амілази, забезпечуючи таким чином її каталітичну активність і разом з тим оберігає від дії протеолітичних ферментів і теплової денатурації.
Велике практичне значення має вплив температури і рН на стабільність амілаз. Швидке руйнування зернової α-амілази за рН 3,3...4,0, наприклад, дає можливість випікати житній хліб з борошна, яке містить надлишок α-амілази, за низьких значень рН, щоб запобігти зайвому декстринюванню крохмалю і утворенню клейких речовин в м'якуші хліба.
За термостабільністю α-амілази різного походження, можна розташувати в наступному ряду в міру зниження їх стійкості до нагрівання: бактерійні амілази → зернові амілази → грибні амілази.
α-Амілаза належить до числа ферментів з досить високою термостабільністю. Мезофільні бактерії продукують фермент, стабільний за температури до 80°С, частіше не вище 70°С. Амілаза термофілів може мати вражаючу стабільність. Так α-амілаза В.licteniformis, що випускається у вигляді комерційного препарату "Термаміл", у присутності 1 М СаС12 і 31,5% крохмалю не втрачає активності за 90°С, а за 100°С час напівінактивації складає більше 3 год.
В насінні рослин є присутніми два типи α-амілази : α-амілаза дозрівання і α-амілаза проростання.
У дозріваючому зерні синтезується α-амілаза дозрівання, яка потім переходить в латентну форму, локалізуючись на мембранах алейронового шару. Перший етап гідролізу крохмалю під час проростання здійснюється цією α-амілазою. І тільки на наступному етапі в роботу включається фермент, що знову синтезується – α-амілаза проростання. Її синтез в клітинах зародка і алейронового шару починається за вологості зерна вище 28%. Дві форми α-амілази насіння злаків відрізняються за термостабільності: α-амілаза дозрівання за 70°С втрачає 50% своєї активності, тоді як α-амілаза проростання за цієї температури тільки трохи знижує свою активність.
Потужним механізмом регуляції швидкості розщеплення крохмальних гранул є система білкових інгібіторів амілаз, широко представлених в рослинах. Інгібітори білкової природи вибірково взаємодіють з амілазами і утворюють неактивні комплекси "амілаза-інгібітор". Високу активність мають інгібітори амілаз картопляного соку. Із зерна пшениці виділений інгібітор з двома активними центрами (двоцентровий). Один активний центр має спорідненість до протеаз і здатний блокувати їх дію. Інший активний центр має спорідненість до амілаз. Таким чином, один інгібітор білкової природи здатний блокувати роботу як протеаз, так і амілаз. У надмолекулярному комплексі, що утворюється, інгібітор виконує своєрідну роль з'єднувальної ланки, пригнічуючи активність ферментів різного механізму дії.
Вдосконалення технології мікробних α-амілаз відбувається в двох основних напрямах: створення препаратів високої термостабільності для гідролізу клейстеризованого крохмалю і препаратів, придатних для гідролізу сирого, неклейстеризованого крохмалю. Другий шлях дасть змогу перейти до низькотемпературного розщеплення крохмалю, що істотно скоротить енерговитрати і спростить апаратурне оформлення процесу гідролізу.
β-Амілаза (α-1,4-глюкан-мальтогідролаза, КФ.3.2.1.2) – фермент екзо-типу, що каталізує послідовне відщеплення мальтози від нередукуючого кінця молекул амілози, амілопектину, амілодекстринів, розриваючи глікозидні зв’язки через один. Фермент розщеплює в крохмалі тільки α-1,4-глікозидні зв'язки. Сирий крохмаль не гідролізує.
На наведеній нижче схемі дія β-амілази на амілозу і амілопектин показана стрілками.
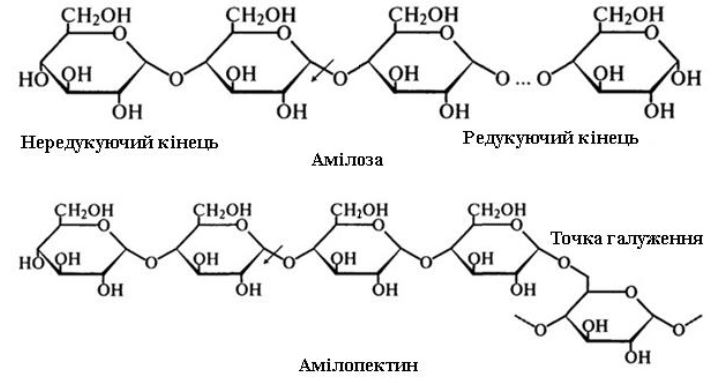
Таким чином, β-амілаза послідовно відщеплює залишки мальтози від нередукуючого кінця до тих пір, доки не зустрінеться точка галуження зі зв’язком α-1,6. Амілоза гідролізується до мальтози повністю, а в результаті дії β-амілази на амілопектин окрім мальтози залишається недоторканою велика, сильно розгалужена серцевина ("кінцевий декстрин"), так як фермент припиняє свою дію за 2...3 залишки глюкози від точок галуження. Нерозщеплені фрагменти амілопектину носять ще назву β-насичених декстринів. В результаті гідролізу крохмалю β-амілазою утворюється 54...58% мальтози і 42...46% насиченого декстрину. Мальтоза в результаті відщепленні переходить в β-форму, що пояснює назву ферменту.
β-Амілазу продукують вищі рослини, мікроорганізми. Фермент міститься в непророслому зерні і солоді злакових культур. Солод злаків довгий час був єдиним джерелом β-амілази, що використовувався на практиці. Зернові β-амілази є найбільш активними за рН 4...6, стабільні за рН 4...8. Оптимальна температура дії 40...50°С, температура стабільності – не вище 60°С. Зернові β-амілази – сульфгідрильні ферменти, їх активність пригнічують важкі метали і окиснювальні агенти.
У зерні β-амілаза є присутньою в активній і латентній формі. В результаті проростання латентна форма активується під дією протеаз.
Багато бактерій, зокрема, бацили синтезують β-амілазу в значних кількостях. Фермент бацил активний за рН 6...7,5, оптимальна температура дії ферменту з різних культур коливається в межах 30...60°С. Бактерійна β-амілаза стабільна за рН 5...9 і температури не вище 55° С.
β-Амілаза рідко використовується як індивідуальний фермент а частіше – в поєднанні з α-амілазою. Комплекс цих ферментів дозволяє розщеплювати крохмаль на 94...96% до мальтози, окрім якої в гідролізаті є присутньою невелика кількість глюкози і низькомолекулярний α-1,6-декстрини.
Глюкоамілаза (α-1,4-глюкан-глюкогідролаза, КФ.3.2.1.3). Глюкоамілаза (γ-амілаза) – екзо-фермент, що каталізує відщеплення β-глюкози від нередукуючого кінця амілози і амілопектину, розщеплюють як амілозу, так і амілопектин до глюкози. Глюкоамілаза розщеплює α- 1,4, α- 1,6 і α-1,3-глікозидні зв'язки, з найбільшою швидкістю – α-1,4.
Глюкоамілаза продукується різними видами цвілевих грибів роду Aspergillus: A.oryzae, A.niger, A.awamory і деякими іншими, наприклад, Rhizopusdelamarn і Rhizopus niveus. Більшість глюкоамілаз – глікопротеїни, вміст вуглеводів – до 35%.
Різні глюкоамілази відрізняються одна від одної швидкістю гідролізу крохмалю, відношенням до температури і рН, і деякими іншими показниками. Грибні глюкоамілази – білки молекулярної маси від 48 до 112 кДа. Максимальна активність проявляється за рН 4,3...5,9 і температури 40...70° С
Глюкоамілази мукорових грибів здатні гідролізувати крохмаль на 95...100%, фермент з пеніцилів аспергилів – на 88...95%. На використанні препаратів грибної глюкоамілази розроблений ферментативний метод одержання глюкози, який отримав широке розповсюдження в Японії.
Інулаза (КФ.3.2.1.7) здійснює ферментативний гідроліз інуліну та інших поліфруктонів по β-1,2-фруктозиднимим зв’язкам, починаючи з β-фруктозидного кінця полімеру. В результаті утворюється фруктоза і єдина молекула глюкози на одну молекулу інуліну. За повного гідролізу інуліну виходить 95% фруктози і 5% глюкози.
Інулін є полімером β-1,2-зв'язаної фруктози, у якого на нередукуючому кінці є один залишок глюкопіранози, приєднаний 1,1-глікозидним зв'язком. Середнє число фруктозних залишків 30...40, молекулярна маса 5...6,5 кДа.
Інулін накопичується як запасний полісахарид у бульбах і коренях рослин, у тому числі у багатьох складноцвітих. Вміст інуліну в % до сухої маси складає: у бульбах топінамбура – 80, коренях цикорію – 75, жоржини – 72, дзвіночків – 45, кульбаби і берізки – 40. Промислове значення має топінамбур, який дає урожай 200...300 ц/га. Сира речовина бульб топінамбура містить приблизно 18% інуліну, що перевищує вміст сахарози в цукровому буряку (11%).
Інулаза міститься в тих же рослинах (топінамбур, цикорій), в яких є присутнім інулін. Активна інулаза продукується Aspergillus awamori BKM-808 за глибинного культивування. Препарат має високу стабільність до Н+-іонів і температури. Оптимум рН дії 4,5; оптимум температури – 65°С.
Отримані із інуліну фруктозні сиропи можуть використовуватись в кондитерській промисловості і дієтичному харчуванні.
Пулуланаза (пулулан-6-глюканогідролаза, КФ.3.2.1.41) гідролізує α-1,6-глікозидні зв'язки в пулулані, глікогені, амілопектині і граничному декстрині, що утворюються в результаті дії на амілопектин α- і β-амілаз. Гідроліз відбувається за ендо-типом, основний продукт розщеплення пулулану – мальтотріоза.
На наведеній нижче схемі дія пулуланази на пулулан показана стрілками:
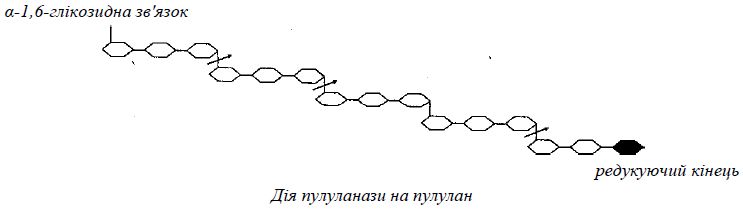
Пулуланаза виділена з різних видів бактерій, переважно з бацил і з актиноміцетів. Фермент проявляє максимальну активність за рН 5...7, деякі штами бацил продукують лужну пулуланазу з оптимумом дії за рН 8,5...9. Оптимальна температура – 45...60° С. Стабільність пулуланаз підвищується у присутності іонів Кальцію.
Пулуланаза разом з іншими амілолітичними ферментами застосовується в технології цукристих продуктів, що отримуються з крохмалю.
Ізоамілаза (глікоген-6-глюканогідролаза, КФ. 3.2.1.68) гідролізує α-1,6-глікозидні зв'язки в субстратах, що галузяться, за винятком пулулану. Слабо гідролізує α-насичені декстрин. Повністю розщеплює глікозидні зв'язки в точках галуження глікогену.
Ізоамілаза виділена з дріжджів, грибів, бактерій. Фермент найбільш активний за рН 3...6 і температури 25...50° С. Використовується у поєднанні з іншими амілазами під час отримання цукристих продуктів з крохмалю.
β-Фруктофуранозідаза (КФ. 3.2.1.26). Інші назви цього ферменту – інвертаза або сахараза.
Для промислового виробництва мають значення ферменти з S.cerevisiae і S. carlsbergensis. β-Фруктофуранозидазу виділяють з дріжджів шляхом автолізу. Цей фермент гідролізує сахарозу за β-фруктозидними зв'язками згідно з рівнянням:
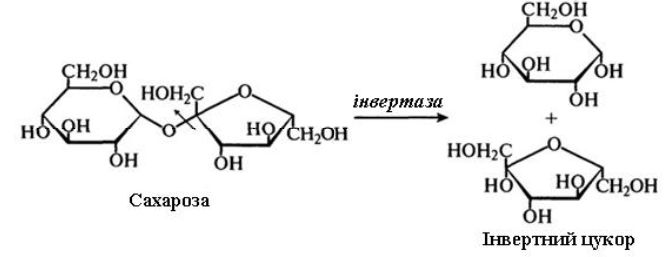
В результаті дії ферменту на сахарозу виходить суміш еквімолярних кількостей α-глюкози і β-фруктози, що дістала назву «Інвертного цукру». Термін «інверсія» означає зміни, що відбуваються в здатності цукру обертати площину поляризованого світла. Це можна виразити наступною схемою:
|
Сахароза + Н20→ |
D(+) -глюкоза + |
D(-) -фруктоза |
|
[a]D=+66,5º |
[а]D=+52,5º |
[a]D=-92,4º |
Інвертаза відщепляє кінцевий невідновлюючий β-1,2-связанний залишок фруктози не лише від сахарози, але і від інших олігосахаридів, частково гідролізує інулін. Під час гідролізу рафінози з однієї її молекули утворюється по одній молекулі лактози і фруктози.
Інвертазу виділяють з культур дріжджів і мікроскопічних грибів. Гриби продукують позаклітинну інвертазу. Активними продуцентами інвертази є гриби родів Aspergillus, Penicillium, Fusarium, Humicola, Malbranchea, Thermomyces. Деякі штами цих грибів осмофільні і сахарозотолерантні, вони здатні рости і розвиватися на середовищах з концентрацією сахарози 20...40%.
Інвертаза знаходить широке застосування в харчовій промисловості. Гідроліз концентрованих розчинів сахарози призводить до утворення солодших сиропів. Точка кипіння інвертованих сиропів вища, а точка замерзання нижча, т. до. за інверсії підвищується осмотичний тиск. Моносахариди, що утворилися в результаті дії інвертази, є більше розчинними, не так легко викристалізовуються з висококонцентрованих сиропів.
Необхідність гідролізу(інверсії) сахарози виникає під час приготування концентрованих цукрових сиропів, морозива, начинки для цукерок.
β-Галактозідаза Фермент, який часто називають лактазою, каталізує реакцію гідролітичного відщеплення нередукуючих залишків β-D-галактози в β-галактозидах, зокрема, в молочному цукрі – дисахариді лактози:
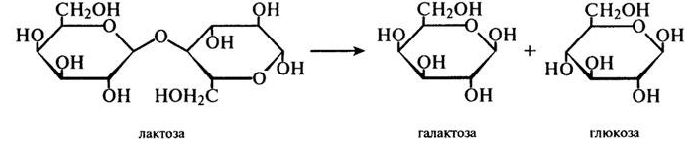
Ферментні препарати лактази, що використовуються в харчовій промисловості, отримують за допомогою різних продуцентів: мікроскопічних грибів (A.oryzae, A.niger), бактерій (Е.coli, Lactobacillus), дріжджів (S.fragilis, S.psedotropicalis). Усі вони мають різний температурний оптимум, який, проте, знаходиться в межах 37...50°С. Оптимум рН цих ферментів також помітно відрізняється: для бактерійних – ≈7,0; для грибних – ≈5,0; для дріжджової лактази – ≈6,0.
Під час гідролізу лактози в незбираному молоці, знежиреному молоці або в концентратах молока оптимальну активність (за нейтрального рН цих субстратів) проявляє дріжджовий фермент; для сироватки і його концентратів – грибний. Причому в знежиреному молоці або сироватці лактоза гідролізується легша, ніж в цілісному, а пастеризовані субстрати гідролізуються легше, ніж непастеризовані.
β-Галактозідаза з E.coli була отримана в кристалічному стані, її молекулярна маса 850 кД. Вона інгібується деякими металами (Сu, Zn). Відновлюючі агенти (цистеїн, сульфід і сульфіт натрію та ін.) активують фермент і здатні долати вплив інгібіторів-металів.
У деяких груп населення (виняток становлять жителі Північної Європи і деяких районів Африки) яскраво виражена непереносимість (інтолерантність) лактози, пов'язана з повним або частковим зникненням у дорослих людей лактазної активності в клітинах кишкового епітелію. У немовлят активність цього ферменту є дуже високою.
Ендополігалактуроназа (КФ.3.2.1.15) і екзополігалактуроназа (КФ.3.2.1.67). Ці два ферменти беруть участь в перетвореннях пектинових речовин разом з іншими пектолітичними ферментами рослинного і мікробного походження.
Ендополігалактуроназа (ендо-ПГ) – фермент, який гідролізує α-1,4-связи в молекулі розчинного пектину (метоксильованої полігалактуронової кислоти), за неврегульованим, довільним механізмом. Зі зростанням ступеня естерифікації полігалактуронової кислоти ступінь і швидкість гідролізу падають, так як для прояву каталітичної активності ферменту потрібні вільні карбоксильні групи. Більшість вивчених ендополігалактуроназ мікроскопічних грибів мають молекулярну масу від 30 до 40 кД. Оптимальні значення рН – 3,8...5,5.
У гідролізі цього типу зв'язку бере участь і інший фермент – екзополігалактуроназа, який послідовно відщеплює молекулу галактуронової кислоти, починаючи з нередукуючого кінця. Ендополігалактуроназа синтезується як грибами, так і деякими видами бактерій. Вони відрізняються за своєю специфічністю до пектинів з різних джерел, кінцевими продуктами реакції, оптимуму рН і іншим властивостям.
Для промислового виробництва ферментних препаратів пектолітичних ферментів, які є комплексними, в якості продуцентів використовують в основному мікроскопічні (цвілеві) гриби, зокрема, гриби роду Aspergillus: A.niger, A.wentii, A.oryzae. Бактерійні ферменти в промислових масштабах не робляться.
Рослинні полігалактуронази є схожими на грибні полігалактуронази. Вони виявлені в широкому спектрі плодів і овочів: помідорах, авокадо, редисці, огірках, яблуках, грушах, цитрусових та ін. Усі вони проявляють активність за природних значень рН плодів.
Застосування препаратів пектолітичних ферментів в промисловості є досить широким. Вони використовуються у виробництві фруктових сокових концентратів і екстрактів, у освітленні соків і вин, у виробництві фруктових і овочевих пюре і нектарів.
Целюлітичні ферменти. Целюлоза є одним з природних полімерів, що найважче піддаються гідролізу. У організмі вищих тварин і людини не синтезуються ферменти, що гідролізують целюлозу. Біодеградацію целюлози здійснюють ферменти мікроорганізмів. Мікрофлора товстого кишечника людини ферментує целюлозу овочів і фруктів повністю. Грубіша целюлоза, що наприклад, входить в препарати харчових волокон, розщеплюється на 0...70%.
У гідролізі целюлози приймають участь три основні види ферментів:
– ендо-β-1,4-глюканази (КФ.3.2.1.4), які каталізують неврегульоване розщеплення целюлозних молекул на великі фрагменти;
– екзо-β-1,4-глюканази, або целюлобіогідролази (КФ.3.2.1.91), в результаті дії яких від нередукуючого кінця целюлозних молекул або їх ферментів відщеплюється целобіоза;
– целобіози, або β-глюкозидази (КФ.3.2.1.21), які каталізують гідроліз целобіози і, з меншою швидкістю, – невеликих целоолігосахаридів, з утворенням глюкози.
Деякі мікроорганізми синтезують екзо-β-1,4-глюкозидазу (КФ.3.2.1.74), під дією якої від нередукуючого кінця целюлозних субстратів відщеплюється глюкоза.
Доведено, що різні ферменти, що гідролізують високовпорядковану целюлозу, діють в синергізмі (рисунок 10.1).
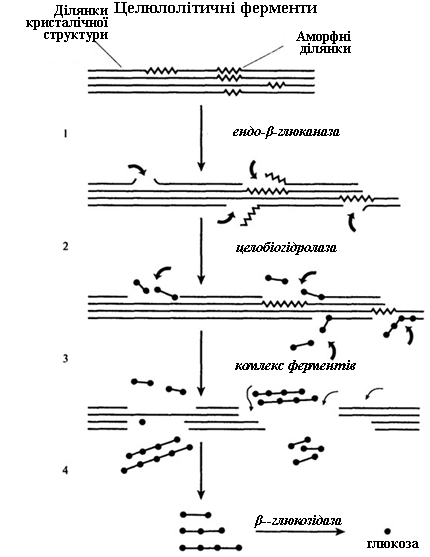
Рисунок 10.1 – Схема ферментативного гідролізу целюлози
Целюлазні комплекси мікроорганізмів і вищих базидіальних грибів включають до 20 ферментних білків, серед яких, як правило, є і ендо-, і екзо-ферменти.
Гідроліз целюлози асоційованими бактерійними целюлазами має місце в рубці жуйних тварин. У рубцевій рідині лише ≈5% целюлаз знаходяться у вільному стані, інша частина представлена асоціатами. У гідролізі целюлози приймають участь різні бактерії, що населяють рубець. За 6...8 год перебування в цьому відділі шлунку целюлоза розщеплюється на 40...50%.
Повнота гідролізу целюлози залежить від ряду факторів, серед яких наступні: ступінь кристалічності субстрату, величина його питомої поверхні, склад ферментативного комплексу, що використовується для гідролізу, і властивості його компонентів.
Нативна целюлоза має дуже міцну структуру і важко гідролізується. Існує обернена залежність швидкості гідролізу від відсотка кристалічності. Для збільшення доступності целюлози для дії ферментів її піддають подрібненню. Таким чином знижується розмір частинок, збільшується питома поверхня субстрату і частка аморфної частини. За сильної механічної дії може навіть знизитись і ступінь полімеризації целюлози. Швидкість гідролізу целюлози прямо пропорційна величині питомої поверхні, вона збільшується в міру зниження розміру частинок і ступеня полімеризації целюлози.
Для гідролізу целюлози використовуються комплексні ферментні препарати, що виділяються з культур мікроскопічних грибів і актиноміцетів і які проявляють ендоглюканазну, целобіогідролазну і целобіазну активність. Окремі компоненти целюлазних комплексів грибів і актиноміцетів проявляють найбільшу активність за рН від 3,7 до 5,5, а комплекси в цілому – за рН 4,5...5,5. Оптимальна температура дії окремих компонентів – від 45 до 80°С, комплексів – 50...60°С. Деякі вищі базидіоміцети синтезують целюлази з оптимумом за рН 3.
Застосування целюлітичних ферментів представляє великий інтерес, оскільки ферментативний гідроліз целюлозовмісних матеріалів (деревина, торф, сільськогосподарські і міські відходи) може забезпечити отримання різних біотехнологічних продуктів (глюкози, етанолу, ацетону, мікробної біомаси).
Протеази (пептидгідролази)
Білок і пептиди розщеплюють ферменти, що об'єднані в підклас пептидгідролаз (КФ. 3.4.). Їх називають також протеазами, протеолітичними ферментами.
Основною реакцією, що каталізується протеолітичними ферментами, є гідроліз пептидного зв'язку в молекулах білків і пептидів :
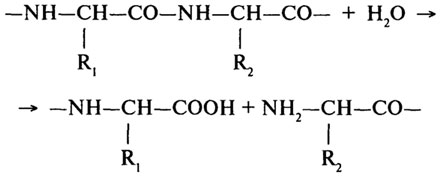
Протеази поділяють на ендо- і екзопептидази. Ферменти першої групи (ендопептидази) можуть гідролізувати глибинні пептидні зв'язки і розщеплювати молекулу білку на дрібніші фрагменти; ферменти другої групи (екзопептидази) не можуть гідролізувати пептидні зв'язки, що знаходяться в середині ланцюга, і діють або з карбоксильного, або з амінного кінця ланцюга, відщеплюючи послідовно одну за іншою кінцеві амінокислоти. У зв'язку з цим екзопептидази піділяють на амінопептидазу, карбоксипептидазу і дипептидазу.
Амінопептидаза (КФ.3.4.11) каталізує відщеплення N-кінцевих амінокислот:
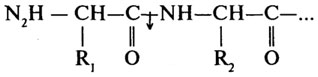
Карбоксипептидаза (КФ.3.4.12) каталізує відщеплення С-кінцевих амінокислот.
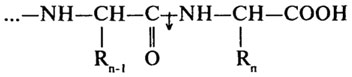
Дипептидаза (КФ.3.4.13) гідролізує дипептиди.
Дипептидилпептидази (КФ. 3.4.14) і пептидилдипептидази (КФ.3.4.15) каталізують відщеплення дипептидов відповідно від N- кінця і від С-конца поліпептидного ланцюга.
В той же час ендопептидази (протеїнази) поділені на підгрупи, починаючи з підпідкласу 3.4.21, в першу чергу на основі каталітичного механізму (будови активного центру); особливості специфічності використовуються тільки для ідентифікації індивідуальних ферментів в межах підпідкласу:
– серинові протеїнази (КФ. 3.4.21), в активному центрі яких функціонує залишок серину і гістидину;
– тіолові (цистеїнові) протеїнази (КФ.3.4.22), містять в активному центрі SH- групу залишку цистеїну;
– кислі (карбоксильні) протеїнази (КФ.3.4.23), в активному центрі містять СООН-групу залишку аспарагінової кислоти;
– металопротеїнази (КФ.3.4.24), містять в активному центрі метал, необхідний для прояву їх каталітичної активності.
Група протеїназ, про механізм дії яких нічого невідомо, віднесена до підпідкласу 3.4.99.
Схема класифікації дана на рисунку 10.2
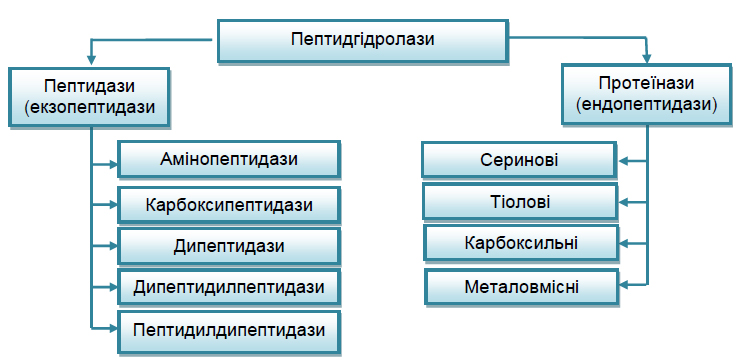
Рисунок 10.2 – Схема класифікації пептидгідролаз
Швидкість ферментативного гідролізу білкових сполук визначається наявністю в них пептидних зв'язків, специфічних для дії ферменту, а також просторовою структурою субстрату.
На доступність пептидних зв'язків гідролізу впливають вторинна, третинна і четвертинна структура білків. Білки можуть мати один або два типи впорядкованих вторинних структур (α-спіральну і β-складчасту), що представлених в різних поєднаннях і охоплюють більш менш значну частину поліпептидного ланцюга. У впорядкованих структурах певні ділянки поліпептидного ланцюга екрановані і недоступні дії ферментів. Чим вище ступінь впорядкованості структури, тим менш білок схильний до протеолізу.
Третинна структура білку, його геометрична форма визначає співвідношення експонованої і екранованої частин молекули (тобто доступною і недоступною протеолізу).
Найменш доступні для протеолізу є молекули з найменшою питомою поверхнею, тобто що наближаються за формою до кулі.
Четвертинну структуру мають білкові молекули, що складаються з субодиниць. Останні можуть бути асоційовані за рахунок ковалентних, іонних і водневих зв'язків. Асоціація субодиниць знижує відносну величину експонованої частини молекули, збільшує її конформаційну стабільність за рахунок внутрішньомолекулярних взаємодій. Асоційовані молекули є менш доступними для дії ферментів, ніж дисоційовані.
Денатурація білків супроводжується розгортанням поліпептидного ланцюга, демаскуванням раніше екранованих груп. Знімаються обмеження доступності субстрату, що зумовлені вторинною, третинною четвертинною структурою. Денатуровані білки гідролізуются швидше і повніше ніж нативні.
Протеази, що мають технологічне значення
Рослинні протеази. Папаїн (КФ.3.4.22.2) і хімопапаїн (КФ.3.4.22.6). Папаїн є найбільш вживаним у виробництві протеолітичним ферментом. Ферменти папаїн і хімопапаїн є істиннми ферментами латексу плодів динного дерева (Carica papaya).
Ці ферменти відносяться до групи тіолових протеїназ, характерною особливістю яких є те, що вони активуються сульфгідрильними сполукими – відновленим глутатіоном, цистеїном.
Папаїн і хімопапаїн отримані в кристалічному стані; їх молекулярна маса 20,7 і 36,0 кД відповідно, а ІЕТ дорівнюють 8,75 для папаїну і 10,1 для хімопапаїну. Оптимальна зона рН для дії папаїну залежить від природи гідролізованого білку і може бути слабокислою, нейтральною або слаболужною. Папаїн має досить широку специфічність. Він переважно гідролізує другий пептидний зв'язок, що знаходиться за карбоксильною групою фенілаланіну.
Ферментні препарати на основі папаїну випускаються з різним ступенем очищення. Можливості їх використання великі: шкіряна промисловість (для видалення волосся і м'якшенні шкур); парфумерія (добавки в креми, лосьйони, зубні пасти); виробництво синтетичних миючих засобів (видалення забруднень білкової природи); медицина (лікування запальних процесів, опіків, тромбозів та ін.); харчова промисловість (виноробство, пивоваріння, виробництво спирту, хлібопечення, сироваріння та ін.).
Фіцин (КФ.3.4.22.3) і бромелаїн (КФ.3.4.22.5). Фіцин виділяють з молочного соку фікусових рослин, наприклад, інжиру (Ficus carica). Так само, як і папаїн, він відноситься до тіоловим протеїназам. Ще один тіоловий фермент – бромелаїн – отримують зі свіжого соку ананасу (Bromeliacea).
Обидва ці ферменти мають подібність з папаїном, проявляють найбільшу активність в нейтральній зоні рН, мають широку специфічність, переважно розщеплюють пептидні зв'язки, утворені позитивно зарядженими амінокислотами.
Використання бромелаїна і фіцину аналогічно використанню папаїну; ці ферменти застосовують також для видалення білкової муті в пиві і для м'якшення м'яса.
Протеолітичні ферменти насіння рослин. У насінні злакових і бобових культур міститься цілий комплекс протеолітичних ферментів, що беруть участь в розщеплюванні запасних білків до амінокислот в процесі проростання насіння. У насінні, що покоїться, стан білкового комплексу характеризується високою стабільністю і автоліз у водних суспензіях виражений слабо.
З насіння пшениці було виділено декілька типів протеолітичних ферментів, що відрізняються за оптимумом рН : кислі протеїнази з оптимумом рН 3,7...4,0; нейтральні протеїнази з оптимумом рН 6,5...7,0; лужні протеїнази з оптимумом рН > 8,0.
З трьох груп протеїназ найбільшої уваги технологів заслуговують нейтральні протеїнази. За активністю вони у декілька разів перевершують кислі і в тісті здатні ефективно розщеплювати білки клейковини. Одна з особливостей нейтральних протеїназ полягає в тому, що вони не розчиняються у водних, сольових і буферних розчинах. Вони міцно зв'язані з білками комплексу клейковини і витягаються за часткового розчинення клейковини в лужному розчині. Таким чином, в дозрілому насінні пшениці нейтральні протеїнази і їх білкові інгібітори утворюють єдиний неактивний комплекс, з'єднаний з клейковиною. Співвідношення активності протеїназ і їх інгібіторів в дозрілому зерні визначає стабільність білкового комплексу, його стійкість в процесі тістоведення.
Нейтральні протеїнази інгібуються хлоридом натрію, фенольними сполуками, ароматичними амінокислотами, продуктами сахаро-аміної реакції (меланоїдинами). Хлорид натрію є обов'язковим компонентом рецептури, так як знижує активність нейтральних протеїназ і відповідно інтенсивність автолізу на 60...70%. В залежності від якості борошна і стану її комплексу клейковини технолог може варіювати час внесення солі і тим самим регулювати інтенсивність протеоліза. Під час переробкиі слабкого борошна необхідно якомога раніше вводити сіль, тоді як для борошна з надмірно міцною клейковиною бажано активізувати протеоліз, і сіль слід вносити на пізніших стадіях.
Протеази тваринного походження. Протеазам тваринного походження належить величезна роль в процесах травлення.
Трипсин (КФ. 3.4.21.4). Трипсин – серинова протеїназа, отримана в кристалічній формі. Молекулярна маса ≈ 23,8 кД. ІЕТ 10,6; оптимум рН дії знаходиться між 7,0...9,0 для білків і синтетичних субстратів. Трипсин проявляє високу специфічність до певних пептидних зв'язків. Він здійснює гідроліз пептидних зв'язків, утворених карбоксильними групами аргініну і лізину.
Високоочищений трипсин застосовується для медичних цілей. Це одна з головних протеаз підшлункової залози, яка у вигляді неочищеного панкреатину знаходить деяке застосування в харчовій промисловості у виробництва гідролізату.
Хімотрипсин (КФ. 3.4.21.1). Хімотрипсин – протеолітичний фермент, що секретується підшлунковою залозою в тонкий кишечник у вигляді неактивного попередника, що називається хімотрипсиногеном.
У активному центрі хімотрипсину міститься залишок гістидину (57), аспарагінової кислоти (102) і залишок серину (195). Молекулярна маса ≈ 225кД. ІЕТ точка 8,3; оптимум рН в межах 7,0...9,0, що узгоджується з природними умовами його дії. Специфічність хімотрипсину полягає в тому, що він переважно гідролізує пептидні зв'язки, утворені ароматичними амінокислотами: тирозином, триптофаном, фенілаланіном.
Цей фермент не застосовується в харчовій промисловості сам по собі, але є складовою частиною комплексних препаратів панкреатину.
Пепсин (КФ. 3.4.23.1). Пепсин виробляється слизовою оболонкою шлунку у вигляді пепсиногену, який перетворюється на активний пепсин під дією НС1 або аутокаталітичним шляхом розщеплення одного пептидного зв'язку. Фермент отриманий в кристалічному виді, його молекулярна маса 35,0 кД, оптимум рН дії 1,8.
Пепсин є кислою (карбоксильною) протеїназой. Його специфічність виражається в переважному гідролізі пептидних зв'язків, утворених групами амінів фенілаланіну і тирозину.
Пепсин має величезне значення як травний фермент, він входить до складу лікарських ферментних препаратів, тонізуючих засобів, жувальної гумки. У харчовій промисловості пепсин використовують для згортання казеїну молока і для розчинення білкової муті в пиві.
Ренін (КФ. 3.4.23.4). Цей фермент, що має багато подібності з пепсином, міститься в соку четвертого відділу шлунку телят. Ренін утворюється з попередника – прореніну. Його молекулярна маса ≈ 40,0 кД, ІЕТ ≈ 4,5. Оптимум рН дії ферменту 3,7.
Ренін є потужною протеазою, що здійснює згортання молока; він є основним компонентом неочищених екстрактів і комплексних промислових препаратів, що використовуються для цієї мети.
Казеїн молока (78% усіх азотистих речовин молока) є фосфопротеїдом, що містить ≈ 6-10% вуглеводів. Він не осаджується іонами Са+2; дія реніну призводить до утворення макроглікопептиду і пара-х-казеїну. Останній осаджується у присутності іонів Са+2 і сприяє осадженню інших фракцій казеїну.
Мікробні протеази. Число мікроорганізмів, що продукують протеази, є надзвичайно великим. Мікробні протеази (грибні і бактерійні) знаходять широке застосування в різних галузях промисловості. Серед них є ферменти, що мають оптимум в нейтральній, кислій і лужній зонах рН; деякі з них проявляють трипсиноподібну дію, інші є пепсиноподобніми ферментами, треті – тіоловими, четверті мають пептидазну активність і т. д.
Найбільшого застосування знайшли лужна серинова протеаза з Bacillus licheniformis, яка використовується в миючих засобах; протеаза з Мусог, яка замінила телячі сичуги у виробництві сиру, а також грибна протеаза з A.oryzae (у комплексі з амілазою), що використовується в хлібопеченні.
Протеази – найбільш важливі промислові ферменти. Рівень споживання препаратів мікробних протеаз ≈40% від усіх використовуваних ферментів.
Субтилізин Карлсберга (КФ.3.4.21.14). Кристалічна форма цього ферменту уперше була отримана в 1952 р., і відтоді субтилізин є найбільш важливою промислово використовуваною мікробною протеазою. Він продукується В. subtilis і В. licheniformis.
Цей фермент складається з одного поліпептидного ланцюга (214 амінокислотних залишків), серед амінокислот відсутній цистеїн. Молекулярна маса ферменту – 27,3 кД, ІЕТ 9,4; оптимум рН 8,0...9,0. Фермент відрізняється високою рН-стабільністю в діапазоні від 5,0 до 11,0.
Субтилізин Карлсберга є сериновою протеїназою, має широку специфічність, прийнятніше гідролізує пептидні зв'язки, утворені ароматичними амінокислотами.
Реніноподібні кислі протеази. Найбільш важливими з практичної точки зору є кислі протеази, що використовуються у виробництві сирів, і які утворюються із культури Mucor pusillus і Mucor mieher.
Нейтральні протеази аспергілових грибів. З культури A.oryzae були виділені дві нейтральних металопротеїнази.
Протеази з різних штамів A. oryzae або з одних штамів, але що ростуть за різних умов, неоднаково впливають на процес хлібопечення навіть у тому випадку, якщо доза ферменту є стандартизованою. Це пояснюється тим, що промислові препарати містять різні кількості протеолітичних компонентів з A.oryzae, а також впливом різноманітних зовнішніх і внутрішніх чинників на протеоліз білків борошна.