Тема 4 Енергетика і напрямок хімічних процесів. Кінетичні Особливості хімічних реакцій
План
4.1 Термодинамічні параметри (функції) стану системи. Енергетика хімічних процесів. Термохімія. Ентропія. Енергія Гіббса. Ентальпійний і ентропійний фактори хімічного процесу
4.2 Термодинамічні розрахунки
4.3 Основні фактори, які впливають на швидкість реакції. Закон дії маси. Механізми хімічних реакцій. Каталіз
4.4 Оборотність хімічних реакцій. Константа хімічної рівноваги. Розрахункові задачі з хімічної кінетики і рівноваги
4.1 Термодинамічні параметри (функції) стану системи
Термодинаміка – це наука, яка вивчає взаємні перетворення різних видів енергії, можливість і напрямок хімічних процесів.
Системою називають будь-яку частину Всесвіту, ізольовану від навколишнього середовища.
Термодинамічна система – це газ, рідина, розчин, тверде тіло, тобто будь-яка сукупність дуже великого числа частинок. Наприклад, газ, поміщений в циліндр – проста термодинамічна система, вода-лід-пара, які знаходяться у рівновазі – складна система.
Макроскопічна система – це система, фізичні властивості якої (маса, об’єм, тиск, температура) можна визначити експериментально. Система називається рівноважною, якщо макроскопічні властивості її постійні за часом.
Фаза – частина системи, однорідна в усіх точках за хімічним складом та фізичними властивостями і відокремлена від інших частин системи поверхнею розділу. Перехід з одного стану в інший називається фазовим. Наприклад:
лід-вода-водняна пара.
Розрізняють наступні види систем:
– ізольовані – не обмінюються із навколишнім середовищем ні масою ні енергією;
– закриті – здійснюють обмін з навколишнім середовищем енергією, але не масою, наприклад розчини в нагрівачах, холодильниках;
– відкриті – обмінюються з навколишнім середовищем і масою і енергією, наприклад, жива клітина, організм.
Стан системи визначається її термодинамічними параметрами або функціями. Основні термодинамічні функції:
U – внутрішня енергія (сукупність всіх видів енергій частинок в системі, це є частина її повної енергії);
Н – теплоємність (ентальпія), Н = U + PV;
S – ентропія: ΔS = QT/T = ΔH/T;
G – ізобарно-ізотермічний потенціал або енергія Гіббса (вільна енергія): G = H – TS;
А – енергія Гемгольца: А = U – TS.
Хімічна термодинаміка вивчає рівновагу хімічних реакцій, фазові переходи і фазовий стан, вивчає наскільки повно відбувається реакція, передбачає можливість здійснення тієї чи іншої реакції.
Перший закон термодинаміки: енергія замкненої системи є величиною сталою. Це означає, що підведена до системи теплота Q витрачається на зміну її внутрішньої енергії ΔU і на виконання роботи А. У математичному вираженні:
Q = ΔU + W; (4.1)
ΔU = U2 – U1, (4.2)
де Q – кількість теплоти, що поглинає система; ΔU – приріст внутрішньої енергії; W – сумарна робота, яку виконала система.
Суму (U + PV) називають ентальпією – Н, тобто
Н = U + PV. (4.3)
Важливо знати зміну ентальпії – ΔН: ΔН = Н2 – Н1. Тоді
ΔН = ΔU + РΔV, або ΔU = ΔН – Р ΔV. (4.4)
Під час ізобарних процесів (Р = const), крім зміни внутрішньої енергії ΔU в системі за рахунок зміни об’єму виконується певна робота, яка дорівнює добутку тиску Р на зміну об’єму системи ΔV: W = PΔV. Отже, тепловий ефект реакції дорівнює: Q = ΔU + PΔV; прийнявши, що U + РV = Н, матимемо, що для ізобарного процесу тепловий ефект реакції дорівнює зміні ентальпії системи:
Qp = Н2 – Н1 = ΔН. (4.5)
ΔН можна визначити за молярною теплоємністю (Дж/(моль·К)) речовини при Р = const Ср: ΔН = Qp = lnСр ΔТ або
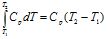 . (4.6)
. (4.6)
Під час ізохорних процесів (V = const, ΔV = 0): Qv = ΔU, вся теплота, підведена до системи витрачається на збільшення U.
Під час ізотермічних процесів (Т = const, U = const, тобто ΔU = 0):
QТ = W = P∙ΔV; dQT = PdV. (4.7)
Для одного моля ідеального газу Р = RT/V, тоді:
W = RTln(V2/V1) = RTln(P1/P2). (4.8)
В адіабатичних процесах (ΔQ = 0): W = -ΔH, тобто робота виконується за рахунок зменшення внутрішньої енергії.
Тепловий ефект реакції – це кількість виділеної (-ΔН) або поглинутої (+ΔН) теплоти.
В хімічній термодинаміці тепловий ефект екзотермічних реакцій позначають зі знаком “-” (система віддає теплоту) або зі знаком “+” (система поглинає теплоту).
Знання теплового ефекту реакції необхідно для розрахунку теплового балансу в хімічному реакторі з метою безпечного проведення реакції для конструювання двигунів, в біохімії, медицині, техніці.
Термохімічні рівняння
Хімічні рівняння, в яких зазначений тепловий ефект реакції, називаються термохімічними рівняннями реакцій, наприклад :
PbO(k) + CO(г) = Pb(k) + CO2(г) ; ΔН = - 64 кДж
Це означає, що при відновленні 1 молю PbO Карбон (ІІ) оксидом виділяється 64 кДж теплоти. Скорочення “к”, “г”, “р” вказують на агрегатний стан речовини. Теплові ефекти реакції вивчає термохімія.
Закони термохімії.
В основі термохімічних розрахунків лежать два основні закони.
- Закон Лавуаз’є-Лапласа: “Тепловий ефект прямої реакції дорівнює тепловому ефекту зворотної реакції з протилежним знаком”.
![]()
- Другий закон термохімії (закон Гесса, 1840 р., російський вчений):” Сумарний тепловий ефект ряду послідовних хімічних реакцій дорівнює сумарному тепловому ефекту будь-якого іншого ряду реакцій з тими самими початковими і кінцевими продуктами реакції”.
Висновок із закону Гесса: тепловий ефект хімічної реакції дорівнює суммі теплот утворення продуктів реакції за вирахуванням суми теплот утворення вихідних речовин з урахуванням коефіцієнтів (ν) в рівнянні реакції. Для реакції
ν1А1 + ν2А2 = ν3А3 + ν4А4.
ΔНр = (ν3![]() + ν4
+ ν4![]() ) - (ν1
) - (ν1![]() + ν2
+ ν2![]() ) (4.9)
) (4.9)
де ![]() - теплота утворення; зміна кількості речовини Δν =(ν3 + ν4)- (ν1 + ν2).
- теплота утворення; зміна кількості речовини Δν =(ν3 + ν4)- (ν1 + ν2).
До термохімічних рівнянь можна застосовувати всі алгебраїчні дії: множення, ділення, перенесення членів рівняння із однієї частини в іншу із протилежним знаком та ін.
Наприклад, розглянемо термохімічне рівняння:
РСІ5(к) + Н2О(г) = РОСІ3(р) + 2НСІ(г); ΔН= -111,4 кДж.
ΔН˚х.р. = ( ![]() + 2
+ 2![]() ) - (
) - ( ![]() +
+ ![]() )= -111,4 кДж.
)= -111,4 кДж.
Для термохімічних даних вибрані стандартні умови: температура 298,16 К (25˚С) і 103,3 кПа (760 мм.рт.ст., 1 атм).
Тепловий ефект (ентальпія) реакції ΔН˚х.р. за стандартних умов, віднесений до 1 моль речовини, називається стандартним тепловим ефектом реакції.
Теплові ефекти реакції можна розрахувати за теплотами утворення, теплотами згорання речовин, теплотами нейтралізації, розчинення та теплотами інших процесів.
Теплотою утворення (![]() ) складної речовини називають стандартний тепловий ефект утворення 1 моль її із простих речовин.
) складної речовини називають стандартний тепловий ефект утворення 1 моль її із простих речовин. ![]() простих речовин дорівнює нулю.
простих речовин дорівнює нулю.
Теплотою згорання речовин називається стандартний тепловий ефект ΔН˚згор. реакції його згорання в кисні до вищих оксидів або до відповідних оксигеновмісних сполук.
В довідникових таблицях вказуються стандартні теплоти утворення ![]() (відомі для багатьох речовин).
(відомі для багатьох речовин).
За теплотами згорання:
ΔН˚х.р= ∑ν ΔНзгор.вих.реч. - ∑ νΔНзгор.прод.реакц. (4.10)
Теплотою нейтралізації називають тепловий ефект взаємодії 1 моля еквіваленту кислоти і 1 моля еквіваленту основи.
Для всіх сильних кислот і основ теплота нейтралізації дорівнює:
Н+ + ОН- = Н2О; ΔН˚х.р = -56,84 кДж.
У випадку слабких кислот або слабких основ тепловий ефект реакції буде нижчий, що зумовлено гідролізом солей, що утворюються.
Теплотою розчинення речовини називається тепловий ефект реакції розчинення 1 моля речовини.
Знаючи тепловий ефект розчинення кристалогідрату і безводної солі, можна обчислити теплоту гідратації цієї солі (тобто теплоту утворення кристалогідрату).
ΔН˚розч.CuSO4 ·5H2O (тепловий ефект розчинення),
ΔН˚розч.CuSO4 (тепловий ефект розчинення і гідратації).
Знаючи теплоти утворення речовин можна обчислити теплові ефекти реакції за їх участю і передбачити їх ймовірний напрямок хоча б за не дуже високих температур. Величина ![]() наближено характеризує стійкість сполук. Наприклад:
наближено характеризує стійкість сполук. Наприклад:
![]() АІ2О3 = -1670,2 кДж/моль
АІ2О3 = -1670,2 кДж/моль
![]() SiO2 = - 859,3 кДж/моль
SiO2 = - 859,3 кДж/моль
Ці сполуки досить стійкі, для їх розкладу необхідні високі температури. Знак “+” біля ![]() свідчить про реакційноздатні сполуки.
свідчить про реакційноздатні сполуки.
Ентропія. Енергія Гіббса. Ентальпійний і ентропійний фактори хімічного процесу
Системи мають тенденцію переходити в стан із найменшою внутрішньою енергією і досягати найбільш стійкого стану. Тому атоми об’єднуються в такі молекули, в результаті яких виділяється найбільша кількість енергії (закон мінімуму енергій). Найбільш ймовірні ті реакції, в ході яких зростає число частинок. Наприклад, для реакції
N2 + 3H2 ![]() 2NH3; ΔН˚ = -91,96 кДж
2NH3; ΔН˚ = -91,96 кДж
а) мінімуму енергії системи відповідає аміак;
б) найбільша ймовірність стану системи – азотно-воднева суміш.
Обидва ці фактори а також результат їх сумісної дії можна виразити кількісно через термодинамічні функції. Іншими словами, самодовільно можуть відбуватися екзотермічні реакції (принцип Бертло). Однак, тепловий ефект реакції не завжди є критерієм, за яким однозначно можна визначити самодовільність того чи іншого процесу, так як самодовільно можуть відбуватися і деякі ендотермічні реакції.
В хімічних реакціях проявляється закон діалектики єдності і боротьби протилежностей. З одного боку, система прагне до впорядкованості (агрегації), до зменшення Н, а з іншого – до невпорядкованості (дезагрегації). Перша тенденція наростає зі зниженням температури, а друга – з її підвищенням.
Для кількісної оцінки відносної ймовірності двох станів системи, або для оцінки ступеня невпорядкованості, введено нову функцію – ентропію S (введена німецьким вченим К. Клаузіусом, 1885 р.). Основною властивістю ентропії є: у будь-якій замкнутій системі, що змінює свій стан у бік рівноваги, ентропія обов’язково зростає. Тобто при будь-яких самодовільних змінах в ізольованій системі ентропія завжди зростає.
Перехід системи з більш впорядкованого в менш впорядкований стан супроводжується збільшенням ентропії. Чим більша ентропія, тим більш невпорядкована система. Рівноважний стан характеризується максимальною невпорядкованістю і найбільшим значенням ентропії.
Зрозуміло, що ентропія зростає в результаті перетворення твердих речовин у рідину, рідин в газ, а також під час розчинення речовин. У всіх цих випадках спостерігається зменшення порядку в розташуванні частинок системи. Навпаки, під час конденсації, кристалізації ентропія речовин зменшується.
Отже, ймовірність стану речовини (газу, рідини, кристалу) можна охарактеризувати як певну властивість системи, яка кількісно виражається ентропією.
Ентропія пов’язана з термодинамічною ймовірність реалізації даного стану системи рівнянням
S = KlnW, (4.11)
де К – константа Больцмана; W – термодинамічна ймовірність, або число можливих мікростанів, які можуть реалізуватись для даного макростану системи.
Ентропію S, Дж/(моль·К) відносять до певної кількості речовини і до певних умов так само, як і ентальпію. З поняття ентропія випливає, що за абсолютного нуля ентропія чистого ідеального кристалу дорівнює нулю. Виходячи з цього, можна обчислити абсолютні значення ентропії за різних температур.
Ентропія залежить від агрегатного стану речовини. S(г) > S(р) > S(к) .
1. Ентропія алотропічних модифікацій тим менша, чим твердіша речовина (алмаз - Sº298 = 2,38; графіт - Sº298 = 5,74);
2. Чим більш складна будова молекули, тим більшою є ентропія (О2 - Sº298 = 205,03, О3 - Sº298 =238,8);
3. Ентропія пару значно більше ентропії рідини (Н2Опар - Sº298 =188,74; Н2Ор - Sº298 = 69,96; Н2Олід - Sº298 = 39,33);
4. Ентропія типових сполук, наприклад солей або за однакового валентного стану елементів, мало відрізняється одна від одної.
Обчислення ΔSº здійснюється аналогічно обчисленню ΔНº за законом Гесса.
Ентальпійний і ентропійний фактори
З розглянутого вище видно, що на перебіг хімічного процесу можуть впливати два фактори: ентальпійний та ентропійний. Самодовільному перебігу процесу сприяє зменшення ентальпії системи, тобто від’ємне значення ΔН . Для хімічних перетворень це, як правило, означає утворення більш складних частинок із менш складних. Крім того, самодовільний перебіг процесу характеризується збільшенням ентропії, або додатним значенням ΔS . Додатна величина ΔS свідчить про тенденцію частинок до подрібнення. Кожна із цих протилежних тенденцій залежить від природи речовини і умов за яких відбувається процес (Т, Р, співвідношення між реагентами та ін.).
В стані рівноваги (обидві тенденції однакові) ΔН = Т ΔS, де ΔН – ентальпійний фактор, а Т ΔS – ентропійний (в кДж/моль). Це рівняння є універсальним (характеризує стан даної системи в умовах рівноваги, коли швидкості протилежних процесів, що в ній відбуваються, стають рівними). Звідси можна розрахувати зміну ентропії в рівноважному процесі: ΔS = ΔН/Т.
Як видно, ентальпійний та ентропійний фактори мають протилежний характер. Можливість перебігу хімічного процесу визначають за енергією Гіббса
ΔG = ΔH – T ΔS.
Це математичний вираз другого закону термодинаміки. За постійної температури і тиску умовою принципової можливості перебігу реакції є від’ємне значення ΔG , тобто ΔG < 0. Якщо реакція відбувається в прямому напрямку, то в зворотному напрямку за даної температури та тиску і концентрації речовини (для реакції в розчинах) вона відбуватися не може, так як в даному випадку ΔG>0 . Ця нерівність є індикатором принципу неможливості процесу.
Коли в системі наступає істинна хімічна рівновага, то ΔGР,Т = 0, так як ΔН = Т ΔS. За цих умов можна розрахувати температуру початку реакції: Т = ΔН/ΔS.
При РV = const напрямок реакції визначається за зміною енергії Гемгольца ΔА. ΔА= ΔU – T ΔS.
Отже, за виразом ΔG = ΔH – T ΔS можна зробити наступні висновки:
1.За низьких температур ΔH > TΔS, напрямок реакції визначається ентальпійним фактором: ΔG ≈ ΔH, тобто знаком ΔH. За високих температур: TΔS > ΔH, тому напрямок визначається ентропійним фактором (прагненням до розриву зв’язків): ΔG ≈ T ΔS. ΔG обчислюють згідно із законом Гесса, так як і ΔH та ΔS, враховуючи, що ΔG простих речовин дорівнює 0. Враховують температуру:
ΔGТ = ΔG298 – ΔS(Т – 298 К).
4.2 Термодинамічні розрахунки
Приклад 1. При сполученні 2,1г заліза із сіркою виділилось 3,77 кДж теплоти. Обчислити теплоту утворення ферум(ІІ)сульфіду.
Розв’язування. Fe(к) + S(к) = FeS(к); ΔНº=?
Врезультаті взаємодії 2,1г Fe виділилось 3,77 кДж
56г Fe х кДж
х = 100,3 кДж/моль
ΔНº= -100,3 кДж/моль
Приклад 2. Які із перерахованих нижче оксидів можуть бути відновлені воднем до вільного металу за 298 К: СаО, NiO ?
Розв’язування.
а) СаО(к) + Н2(г) = Са(к) + Н2О(г)
ΔGº298 = ΔGº ![]() – ΔGºСаО = -228,6 – (-604,2) = 375,6
– ΔGºСаО = -228,6 – (-604,2) = 375,6
Таким чином ΔGº298 > 0, отже, реакція неможлива.
б) NiО(к) + Н2(г) = Ni (к) + Н2О(г)
ΔGº298 = ΔGº ![]() – ΔGº NiО == -228,6 – (-211,6) =-17
– ΔGº NiО == -228,6 – (-211,6) =-17
Таким чином ΔGº298 < 0, отже, реакція можлива.
4.3 Основні фактори, що впливають на швидкість хімічних реакцій
Швидкість і механізм хімічних реакцій вивчає хімічна кінетика. Важливість цього питання можна показати на слідуючих прикладах:
2Н2(г) + О2(г) = 2Н2О(г); ΔGº298 =-457,2 кДж/моль
Не дивлячись на те, що ΔGº298 < 0, внаслідок низької швидкості реакція практично не відбувається.
2NО(г) + О2(г) = 2NО2(г); ΔGº298 =-70 кДж/моль
Ця реакція відбувається за кімнатної температури практично миттєво.
Основна задача хімічної кінетики – можливість керування хімічним процесом, з метою прискорення або сповільнення швидкості реакцій, досягнення максимально високого виходу продукту реакції.
В хімічних реакціях можуть приймати участь газоподібні, рідкі або тверді речовини. Тому розрізняють гомогенні та гетерогенні реакції.
Гомогенні реакції відбуваються в однорідному середовищі (газові суміші, рідкі розчини).
Гетерогенні реакції – в неоднорідному середовищі, коли речовини знаходяться в різних фазах. Для гомогенних процесів, що відбуваються без зміни об’єму, швидкість хімічної реакції визначають як зміну концентрацій реагуючих речовин або продуктів реакції за одиницю часу. Для гетерогенних реакцій швидкість визначають як число елементарних актів реакції, що відбуваються в одиницю часу на одиниці поверхні розділу фаз. Середня швидкість реакції дорівнює: V = -ΔC/Δτ, знак “-“ означає, що концентрація вихідних речовин зменшується з часом.
Миттєва швидкість реакції виражається першою похідною концентрації “С” за часом τ:
![]() , С моль/л (4.12)
, С моль/л (4.12)
Залежність концентрації вихідної речовини від часу можна представити графіком:
|
|
в кожний даний момент часу (наприклад, τ) миттєва швидкість реакції дорівнює тангенсу кута нахилу кривої V = tgα. Істинна швидкість реакцій дорівнює тангенсу кута нахилу дотичної до кривої залежності концентрації від часу: V = tgα. |
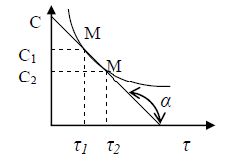
Швидкість хімічної реакції залежить:
- Від природи реагуючих речовин. В даному випадку велику роль відіграють як внутрішньомолекулярні (хімічні) зв’язки, так і міжмолекулярні (ван-дер-ваальсові) сили. Реакції:
Н2 + F2 ![]() 2HF (відбувається в темноті, на холоді, з вибухом)
2HF (відбувається в темноті, на холоді, з вибухом)
H2 + Cl2 ![]() 2HCl (відбувається над дією сонячного світла)
2HCl (відбувається над дією сонячного світла)
H2 + I2 ![]() 2HI (проходить тільки при нагріванні)
2HI (проходить тільки при нагріванні)
2. Від стану реагуючих речовин. В даному випадку дуже важливу роль відіграє агрегатний стан речовини, ступінь дисперсності, чим він більший, тим більшою буде поверхня контакту.
3. Від середовища, в якому відбувається реакція. Так на швидкість реакції у розчинах великий вплив має природа розчинника, можливість утворення водневих зв’язків між молекулами розчинника та розчиненої речовини.
4. Від зовнішніх умов. В даному випадку велику роль відіграють температура, тиск, наявність каталізатора.
5. Від концентрації. Чим вища концентрація, тим більше частинок речовини знаходиться в одиниці об’єму, тим більша ймовірність співударів, що призводить до підвищення швидкості реакції.
Ця залежність визначається законом дії маси: за постійної температури швидкість хімічної реакції прямопропорційна добуткові концентрацій реагуючих речовин, які входять до рівняння хімічної реакції у степенях, що дорівнюють їхнім стехіометричним коефіцієнтам (К. Гульберг, П. Вааге – норвежські вчені; основи для цього закону заложив російський вчений М.М. Бекетов).
Для реакції типу:
аА + вВ ↔ сС + dD
закон дії маси виражається співвідношенням:
![]() (пряма реакція), (4.13)
(пряма реакція), (4.13)
![]() (зворотна реакція), (4.14)
(зворотна реакція), (4.14)
де ![]() і
і ![]() – швидкість реакцій , k1 і k2 – коефіцієнти пропорційності або сталі швидкості реакцій. “k” – це швидкість реакції коли концентрація реагуючих речовин дорівнює 1 моль/л.
– швидкість реакцій , k1 і k2 – коефіцієнти пропорційності або сталі швидкості реакцій. “k” – це швидкість реакції коли концентрація реагуючих речовин дорівнює 1 моль/л.
k залежить від природи реагуючих речовин і температури, наявності каталізаторів, і не залежить від концентрації; [A], [B], [C], [D] –концентрації речовин в моль/л; а, b, c, d – стехіометричні коефіцієнти в рівнянні реакції. У вираз швидкості реакції не входять концентрації твердих речовин.
Механізм хімічних реакцій. Розрізняють елементарні (одно стадійні) і неелементарні (складні, багатостадійні) реакції, при чому, більшість реакцій – багатостадійні.
Число молекул, які приймають участь в елементарному акті хімічної реакції, називається молекулярністю реакції. Це завжди позитивне число, ціле: 1, 2, дуже рідко 3. Розрізняють:
а) мономолекулярні
N2О4 ![]() 2NО2 Ư = k [N2О4]
2NО2 Ư = k [N2О4]
A ![]() B + C Ư = k [A]
B + C Ư = k [A]
б) бімолекулярні
H2 + I2 ![]() 2HI Ư = k [H2][ I2]
2HI Ư = k [H2][ I2]
A + B ![]() C Ư = k [A] [B]
C Ư = k [A] [B]
в)тримолекулярні (зустрічаються досить рідко)
2Н2 + О2 ![]() 2Н2О Ư = k [H2]2[О2]
2Н2О Ư = k [H2]2[О2]
2A + B ![]() C Ư = k[A]2[B]
C Ư = k[A]2[B]
Чотирьохмолекулярні реакції – невідомі.
Сума показників ступенів в кінетичному рівнянні є загальним (сумарним) порядком реакції. Він визначає залежність швидкості реакції від концентрації. Порядок і молекулярність реакції не завжди співпадають. Порядок реакції може бути:
- нульовим, коли швидкість реакції не залежить від концентрації в об’ємі (відбувається на поверхні різних речовин);
- першим: CuO + H2 → Cu + H2O, Ư = k [H2], а молекулярність реакції дорівнює 2;
- другим, коли сума показників ступенів дорівнює двом:
- Ư = k[A]2 або Ư = k [A] [B].
- а також дробним – у випадку багатостадійних реакцій, коли найбільш повільні із них визначають швидкість всієї реакції, а швидкість цих елементарних стадій приблизно однакова, а порядок різний.
Наприклад реакція
2N2О5 → 4NО2 + О2
є реакцією першого порядку. Вона відбувається у дві стадії. Перша стадія – повільна:
N2О5 → N2О3 + О2
і друга:
N2О3 + N2О5 → 4NО2.
Тоді, не дивлячись на те, що це бімолекулярна реакція Ư = k [N2О5]2 , вона відноситься до реакції першого порядку і її швидкість визначається найбільш повільною стадією: Ư = k[N2О5] із рівняння реакції
N2О5 ![]() N2О3 + О2.
N2О3 + О2.
Вплив температури на швидкість реакції
Температура є важливим фактором, що впливає на швидкість реакції. Підвищення температури значно прискорює хімічний процес. Наприклад, для синтезу 15% води за 20ºС необхідно 54 млрд. років, а за 500ºС – 50 хвилин, при 700ºС – реакція відбувається миттєво. Встановлена наступна закономірність (правило Вант-Гоффа) залежності швидкості реакції від температури: з підвищенням температури на кожні 10ºС швидкість реакції зростає в 2-4 рази (це правило для сучасної хімії представляє лише історичний інтерес, так, як має наближений характер і справедливе тільки для реакцій з малою енергією активації).
Вплив нагрівання на швидкість реакції визначається, в основному, зростанням константи швидкості реакції k, так як температура на концентрацію не впливає. Цей вплив характеризується так званим температурним коефіцієнтом швидкості реакції (γ),який дорівнює відношенню константи швидкості при температурі (t1+10) до константи швидкості при температурі t1:
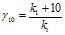 , (4.15)
, (4.15)
звідси, за правилом Вант-Гоффа:
![]() , (4.16)
, (4.16)
де ![]() – швидкість реакції за початкової температури,
– швидкість реакції за початкової температури, ![]() – швидкість реакції при кінцевій температурі; γ – температурний коефіцієнт швидкості, який показує, у скільки разів збільшується швидкість реакції з підвищенням температури на кожні 10ºС.
– швидкість реакції при кінцевій температурі; γ – температурний коефіцієнт швидкості, який показує, у скільки разів збільшується швидкість реакції з підвищенням температури на кожні 10ºС.
Подібно до нагрівання швидкість реакції зростає під дією світла (видимого, ультрафіолетового), реакції синтезу при цьому називаються фотосинтезом, а розкладу – фотолізом; під впливом γ- проміння – радіолізом.
Енергія активації
Не кожне зіткнення між молекулами призводить до утворення нової речовини. В хімічну взаємодію вступають тільки активні молекули, які володіють достатнім запасом енергії, для того, щоб подолати сили відштовхування (енергетичний бар’єр) між електронними оболонками. Звідси, молекули, які володіють максимальною енергією, називаються активними, а додаткова енергія, яку необхідно їм надати для переведення в активний стан – енергією активації (в кДж/моль). Вона завжди менша, ніж енергія розриву хімічного зв’язку.
Константа швидкості реакції k пропорційна не загальному числу молекул в одиниці об’єму системи, а тільки числу активних із них. Тому, чим вища концентрація активних молекул в системі, тим більша k, і тим швидше відбувається хімічний процес. Залежність константи швидкості реакції від температури (від енергії активації) виражається рівнянням Арреніуса (теорія активних зіткнень):
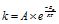 , (4.17)
, (4.17)
де k- константа швидкості; А – перед експоненціальний множник;
е – основа натурального логарифма (е=2,718); Еа – енергія активації;
R – універсальна газова стала; Т – температура; ![]() = а – частка активних молекул.
= а – частка активних молекул.
Звідси, константа швидкості реакції тим більша, чим менша енергія активації і чим вища Т.
Сучасною теорією хімічної кінетики є теорія перехідного стану (активованих комплексів). Так, для реакції типу: АВ + С →АС + В спочатку утворюється активний комплекс (А…В…С), а потім – кінцеві продукти:
АВ + С→ А…В…С→ АС + В
Таким чином, процес розглядається з точки зору структури, енергетичних змін в системі з урахуванням перерозподілу зв’язків та їх послаблення. Різниця між енергією активованого комплексу і середньою енергією молекул вихідних речовин і є енергія активації. Різниця енергій активації прямої (![]() ) і зворотної (
) і зворотної (![]() ) реакцій дорівнює тепловому ефекту реакції: ΔН=
) реакцій дорівнює тепловому ефекту реакції: ΔН=![]() –
– ![]() . Якщо ΔH > 0 (ендотермічна реакція), то
. Якщо ΔH > 0 (ендотермічна реакція), то ![]() >
> ![]() ; якщо ΔН < 0 (екзотермічний процес), то
; якщо ΔН < 0 (екзотермічний процес), то ![]() <
< ![]() . Коли під час розпаду активованого комплексу на продукти реакції енергії виділяється більше, ніж витрачено на активацію молекул вихідних речовин, то реакція буде екзотермічною, а якщо менше – ендотермічною.
. Коли під час розпаду активованого комплексу на продукти реакції енергії виділяється більше, ніж витрачено на активацію молекул вихідних речовин, то реакція буде екзотермічною, а якщо менше – ендотермічною.
Розглянемо реакцію:
H2(г) + J2(г) = 2HJ(г); ΔНº= -17 кДж
Ця реакція може відбуватися такими двома способами:
а) шляхом дисоціації молекул Н2 і J2 на атоми із наступною їх взаємодією і утворенням молекули НJ;
б) активні молекули Н2 і J2 при зіткненні об’єднуються в проміжний активний комплекс Н2 . . . J2, де зв’язки Н – Н і J – J значно послаблюються і при розпаді Н2 . . . J2 синхронно рвуться зв’язки Н – Н і J – J, а зв’язки Н – J утворюються, що схематично зображають так:
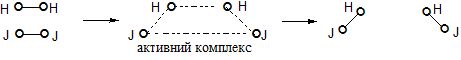
Розраховано, що у випадку а) енергія активації дорівнює 571 кДж/моль, а для випадку б) – 168 кДж/моль .
Висновок: шлях реакції через проміжний активний комплекс енергетично більш вигідний, ніж шлях через повний розрив зв’язків молекул, які вступають в реакцію. Більшість реакцій відбувається через стадію утворення проміжних активних комплексів. Отже, енергія активації – це енергія, яка необхідна для переведення реагуючих речовин в стан активного комплексу.
В реакції можуть приймати участь атоми, молекули, радикали або йони. В зв’язку із цим розрізняють прості, йонні та радикальні реакції.
Простими називають реакції, які відбуваються між молекулами:
H2 + I2 ![]() 2HI
2HI
NО + СІ2 ![]() NОСІ2
NОСІ2
Енергія активації складає 150 – 450 кДж/моль.
Йонними є реакції, які протікають за участі йонів – заряджених частинок. Енергія активації складає 0 – 80 кДж/моль. Утворення йонів може відбуватися при дисоціації речовини, під дією електророзряду, наприклад, випромінюванні високих енергій і т. д. Наприклад, дія γ-проміння:
Н2О + γ → Н2О+ + ē;
Н2О + ē → Н2О-
СН4 + γ → СН4+ + ē
Молекулярні йони є надзвичайно реакційноздатними і за звичайних умов існують лише мільйонні долі секунди.
Радикальні реакції – реакції, які відбуваються через проміжне утворення вільних радикалів (останні можна уявити як осколки молекул, що містять непарні електрони).
Сюди відносять і вільні атоми. Енергія активації таких реакцій складає 0 – 40 кДж/моль.
Ланцюгові реакції ( М.М. Семенов, С.Н. Хіншенвульд). Радикальні реакції протікають за ланцюговим механізмом. Їхня особливість полягає в тому, що один первинний акт активації призводить до перетворення великого числа молекул вихідних речовин. Наприклад реакція:
H2 + СІ2 ![]() 2HСІ
2HСІ
відбувається під час нагрівання або опромінення сонячним світлом. За рахунок поглинання квантів світла (hν) молекула хлору дисоціює на вільні атоми – радикали Хлору СІ2 ![]() СІ· + · СІ, які потім реагують з молекулами водню, утворюючи молекулу НСІ і атом – радикал Н, останній взаємодіє з молекулою Cl2 p з утворенням НСl та радикалу СІ· і т. д.
СІ· + · СІ, які потім реагують з молекулами водню, утворюючи молекулу НСІ і атом – радикал Н, останній взаємодіє з молекулою Cl2 p з утворенням НСl та радикалу СІ· і т. д.
Н2 + СІ → НСІ + Н·
Н·+ СІ2 → НСІ + СІ· та ін.
Вище були наведені приклади нерозгалужених ланцюгових реакцій (утворювався один активний центр). На кожний квант світла, що поглинається утворюється до 100 тисяч молекул НСІ. За ланцюговим механізмом відбуваються реакції вибуху, горіння, окиснення вуглеводнів, полімеризації, ядерні реакції.
Прикладом розгалуженої ланцюгової реакції (утворення двох або більше вільних радикалів) є окиснення Гідрогену.
Спостерігається лавиноподібне проходження процесу з утворенням величезного числа вільних радикалів:
Н + О2 → ОН· + ·О·; ОН· + Н2 → Н2О + Н·;
О + Н2 → ОН· + Н· і т.д.
Каталіз
Каталіз – це селективне прискорення хімічної реакції за наявності речовин-каталізаторів. Він відіграє важливу роль хімічній технології для вирішення проблем прискорення або сповільнення швидкості хімічних реакцій.
Каталізатори – речовини, які змінюють швидкість хімічної реакції, приймаючи участь у проміжних стадіях, але самі при цьому не витрачаються. Каталізатори, які прискорюють швидкість реакції називаються позитивними, а ті, які сповільнюють – негативними (інгібітори, стабілізатори, антикаталізатори). Вони широко використовуються в промисловості і в техніці. Промотори – речовини, які підвищують активність каталізаторів; каталітичні отрути – речовини, які пригнічують дію каталізаторів (знижують або зовсім знищують їх активність).
Розрізняють гомогенний та гетерогенний каталіз.
Гомогенний каталіз – реакційна суміш і каталізатор утворюють одну фазу (газ або рідина). Наприклад:
SO2 + NO2 → SO3 + NO; 2NO + O2 → 2NO2; NO –каталізатор.
Каталіз пояснюється утворенням проміжних сполук за участі каталізатора (активованих комплексів), наприклад А + В → АВ; А + К → АК, де К – каталізатор; АК + В → АКВ → АВ + К; при цьому змінюються термодинамічні параметри: ΔG, ΔH, ΔS, тим самим змінюється константа швидкості і швидкість реакції. Частіше всього каталізатор суттєво знижує Еа процесу і значно підвищує швидкість реакції. (в 102 – 1020 разів) при А=cоnst; але є реакції в яких Еа майже не змінюється, а сильно зростає (на декілька десятитисячних порядків) передекспоненціальний множник А (див. рівняння Арреніуса).
Гетерогенний каталіз характеризується тим, що реакційна суміш і каталізатор утворюють різні фази. Наприклад:
2SO2 + O2 ![]() 2SO3
2SO3
Під час гетерогенного каталізу також утворюються проміжні поверхневі активовані сполуки на активних центрах поверхні каталізатора. Тому, дуже важливе значення в цьому випадку має поверхня контакту, явища адсорбції та хемосорбції.
4.4 Оборотність хімічних реакцій. Константа хімічної рівноваги
Ряд хімічних процесів відбуваються в одному напрямку, такі реакції називаються необоротними. Наприклад:
2КСІО3 → 2КСІ + 3О2
Pb(N3)2 → Pb + 3N2
і проходять, доки не витратиться одна із реагуючих речовин, тобто до кінця.
Інші реакції (їх більшість) відбуваються як у прямому так і в зворотному напрямках, тобто є оборотними. Такі реакції не проходять до кінця. Наприклад:
СаСО3 ![]() СаО + СО2
СаО + СО2
В оборотному процесі, коли швидкість прямої і зворотної реакції стають однаковими, наступає момент рівноваги (концентрації речовин не змінюється і називаються рівноважними концентраціями за даних умов). Але ні пряма, ні зворотна реакція не припиняються, тобто процес відбувається в обох напрямках із однаковими швидкостями. Тому хімічна рівновага є рівновагою динамічною (рухомою).
Для оборотної реакції:
аА + вВ ↔ сС + dD
швидкість реакцій відповідно дорівнює
![]() і
і ![]() (4.18)
(4.18)
в момент рівноваги ![]() =
= ![]() :
:
![]() =
= ![]() або
або ![]() , (4.19)
, (4.19)
де Кс – константа хімічної рівноваги, а наведене рівняння, де фігурують рівноважні концентрації [A], [B], [C], [D] – одне із виразів закону дії маси для реакцій із простим механізмом.
Константа хімічної рівноваги визначає глибину проходження процесу в момент рівноваги. Чим більше К, тим повільніше взаємодіють речовини. Кс залежить від природи реагуючих речовин і від температури, але не залежить від концентрації реагуючих речовин. Крім концентраційної (кінетичної) константи Кс є термодинамічна константа хімічної рівноваги – Ка, яка містить активні концентрації (а= fс, де f – коефіцієнт активності) або активності а. У вираз константи рівноваги не входять концентрації твердих речовин. Наприклад:
СО2(г) + С(к) = 2СО(г); 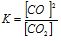
Для визначення константи рівноваги реакції взаємодії між газоподібними речовинами користуються формулою:
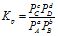 ,
,
в якій реагенти подані рівноважними парціальними тисками Рі.
Кр = КсRTΔν, якщо Δν= 0, Кр = Кс.
Константа хімічної рівноваги пов’язана зі зміною стандартного ізобарно-ізотермічного потенціалу хімічної реакції: ΔGº=-RT lnK.
Якщо врахувати, що ΔG = ΔH – T ΔS, то після деяких перетворень отримаємо:
![]() ; ln (K2/K1) =
; ln (K2/K1) = 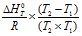 (4.20)
(4.20)
Для екзотермічної реакції із підвищенням температури константа рівноваги зменшується, а для ендотермічних реакцій зі зростанням температури Кс збільшується. Якщо ΔH прямує до нуля, то температура практично не впливає на константу хімічної рівноваги.
Висновки:
- Більш негативним значенням ΔGº відповідає більше значення Кс , тобто в рівноважній системі (суміші) переважають продукти взаємодії, і, навпаки, при ΔGº > 0 – в рівноважній суміші переважають вихідні речовини.
- Константа рівноваги дуже чутлива до зміни температури.
- Вплив на константу рівноваги природи реагуючих речовин визначає її залежність від ентальпійного та ентропійного факторів.
Знаючи концентрацію вихідних речовин і величину Кс, можна обчислити рівноважні концентрації всіх реагуючих речовин за умов хімічної рівноваги. І, навпаки, за рівноважними концентраціями легко обчислити Кс і вихідні концентрації взятих для реакції речовин.
При зміні температури, тиску або концентрації реагуючих речовин, рівновага порушується, при цьому змінюються рівноважні концентрації всіх речовин, які беруть участь у реакції. В результаті переважного проходження реакції в одному із можливих напрямків встановлюється новий стан хімічної рівноваги, який відрізняється від попереднього. Процес переходу від одного рівноважного стану до іншого, називається зміщенням хімічної рівноваги. Визначити напрямок, в який зміститься хімічна рівновага можна за принципом Ле Шателье: якщо на систему, яка знаходиться в рівновазі, подіяти ззовні (змінити температуру, тиск чи концентрацію реагуючих речовин), то рівновага зміститься в бік тієї реакції, яка послаблює цю дію.
Вплив зміни концентрації на хімічну рівновагу: збільшення концентрації одного із компонентів рівноважної системи призводить до зміщення хімічної рівноваги в бік підсилення тієї реакції, за якої відбувається переробка цього компоненту; зменшення концентрації одного із компонентів системи зміщує рівновагу в бік утворення останнього.
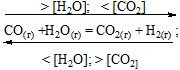
В даному випадку практичне значення відіграє питання виходу водню, тому задача полягає в тому, щоб максимально переробити вихідні речовини.
Вплив температури на хімічну рівновагу
Нагрівання зміщує рівновагу в бік ендотермічного процесу, охолодження, навпаки, - в бік екзотермічного процесу.

В реакціях, які відбуваються практично без теплових ефектів, зміна температури не призводить до зміщення рівноваги (наприклад, реакції етерифікації). В даному випадку підвищення температури призводить лише до більш швидкого встановлення тієї ж рівноваги, яка була б досягнута в даній системі і без нагрівання, але за більш довший проміжок часу.
Вплив тиску на хімічну рівновагу
В лівій частині рівняння в сумі є 4 молі газів, а в правій – 2 молі. При збільшенні тиску рівновага зміщується в бік реакції, яка відбувається зі зменшенням об’єму (вправо), а зменшення тиску – в бік реакції, яка відбувається зі збільшенням об’єму (вліво).
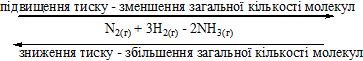
Якщо в ході оборотної реакції загальне число молекул не змінюється, то зміна тиску не впливає на стан хімічної рівноваги.